Angelmanův syndrom (AS) je vzácná neurogenetická porucha, která postihuje přibližně jednoho z 15 000 lidí – asi 500 000 jedinců po celém světě. Děti a dospělí s AS mají obvykle problémy s rovnováhou, poruchy motoriky a oslabující záchvaty. Někteří jedinci nikdy nechodí. Většina nemluví. Přerušené spánkové cykly také mohou být vážnou výzvou pro jednotlivce a správce. Jedinci s AS vyžadují nepřetržitou péči a nejsou schopni žít samostatně. Mají normální délku života. Toto je život dnes pro lidi žijící s Angelmanovým syndromem, ale naděje je tady. Vědci se domnívají, že AS má největší potenciál k vyléčení ve srovnání s jinými neurogenetickými poruchami a FAST (Foundation for Angelman Syndrome Therapeutics) má dobře připravený plán, jak toho dosáhnout.
Typy způsobují testy & Diagnostické zdroje
Typy Angelmanova syndromu
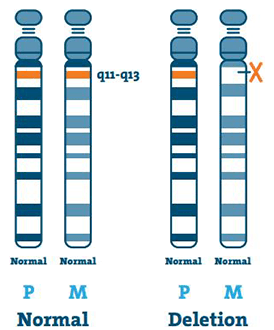
Smazání ( 65-75%)
DNA (deoxyribonukleová kyselina) je hlavní složkou chromozomů. Obsahuje náš jedinečný genetický kód. Většina osob s AS chybí kousek DNA v oblasti 15q11-13 na mateřském chromozomu 15.
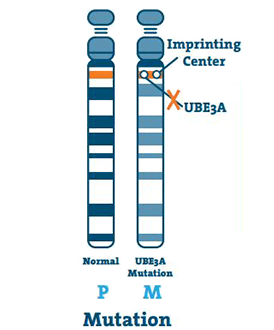
Mutace (5 -11%)
K tomu dochází, když je v DNA genu UBE3A malá abnormalita. Mutace může nastat kdekoli na genu.
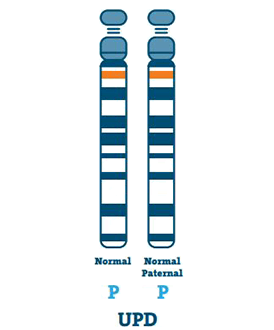
Uniparental Disomy (3-7%)
Jednotlivec s UPD má dvě kopie chromozomu 15 od svého otce, namísto jednoho po jednom od otce a matky. UPD se obvykle stane, pokud ve vejci není chromozom 15.
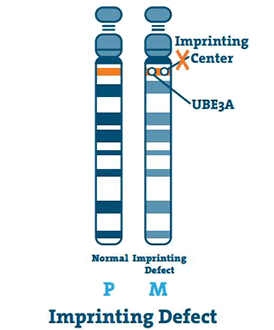
Vada otisku (< 3%)
Centrum pro otisk je malý úsek DNA nacházející se v oblasti q11-13 chromozomu. Ve vzácných případech je matčin chromozom 15 prázdný a střed kopíruje otcův chromozom 15. Toto je vada otisku.
Vlastnosti
Typické vlastnosti AS nejsou obvykle patrné na narození. Lidé s touto poruchou mají potíže s krmením jako kojenci a znatelný opožděný vývoj kolem 6-12 měsíců věku. Potřebují intenzivní terapie, které jim pomohou rozvíjet funkční dovednosti. AS ovlivňuje všechny rasy a obě pohlaví. Často bývá diagnostikována nesprávně jako autismus nebo dětská mozková obrna.
Vlastnosti chování
Lidé s Angelmanovým syndromem mají některé charakteristické rysy chování, včetně šťastného chování, charakterizovaného častým smíchem, úsměvem a vzrušivostí. Mnoho lidí s AS je fascinováno vodou a velmi se těší z aktivit, jako je plavání a koupání.
Příčiny Angelmanova syndromu
Angelmanova syndrom je porucha jednoho genu způsobená ztrátou funkce v genu UBE3A na mateřském 15. chromozomu. Lidé mají dvě sady chromozomů – jednu zděděnou od matky a jednu od otce. U typické osoby je mateřsky zděděný gen UBE3A aktivní, zatímco kopie genu zděděného po otci je umlčena v neuronech v našich mozcích – což je jev známý jako otisk. U lidí s AS tento mateřský gen nedělá svou práci a to má dopad na jejich Messenger RNA (mRNA).
![]()
Co je mRNA?
mRNA je FedEx těla. Naše DNA používá mRNA jako doručovací službu k zasílání plánů do továren na sestavování proteinů našich buněk. Lidé s AS mají mutaci, deleci nebo jinou vadu ve svém genu UBE3A, která přerušuje tuto doručovací službu. Výsledkem je, že jejich neurony nevytvářejí žádný funkční protein UBE3A, a právě to spouští příznaky AS. Tento protein nám pomáhá chodit, mluvit a plnit další každodenní úkoly.
![]()
Je to genetické?
Ve většině případů není Angelmanov syndrom zděděn – zejména ty, které jsou způsobeny delecí nebo UPD. Místo toho k těmto genetickým změnám dochází jako náhodné události během formování reprodukčních buněk nebo v časném embryonálním vývoji.
Testy & Diagnostika
Zjistit o kritériích, která lékaři používají k určení, zda má jedinec Angelmanova syndrom, a přečíst si o dostupných testech a běžných chybných diagnózách.
![]()
Testování
![]()
Diagnostika

Připojte se k naší komunitě
Spojte se s dalšími lidmi, kteří mají dítě nebo někoho milovaného s Angelmanovým syndromem.
