Angelmanin oireyhtymä (AS) on harvinainen neurogeneettinen häiriö, joka vaikuttaa noin yhteen 15 000 ihmisestä – noin 500 000 yksilöön maailmanlaajuisesti. Lapsilla ja aikuisilla, joilla on AS, on tyypillisesti tasapainohäiriöitä, motorisia häiriöitä ja heikentäviä kohtauksia. Jotkut ihmiset eivät koskaan kävele. Useimmat eivät puhu. Myös häiriintyneet unisyklit voivat olla vakava haaste yksilölle ja hoitajalle. Henkilöt, joilla on AS, tarvitsevat jatkuvaa hoitoa eivätkä pysty elämään itsenäisesti. Heillä on normaali elinajanodote. Tämä on nykyään Angelmanin oireyhtymässä elävien ihmisten elämä, mutta toivo on täällä. Tutkijat uskovat, että AS: lla on suurin potentiaali parantua verrattuna muihin neurogeneettisiin häiriöihin, ja FAST: lla (Foundation for Angelman Syndrome Therapeutics) on suunnitelma tämän saavuttamiseksi.
Tyypit aiheuttavat testejä & Diagnoosiresurssit
Angelmanin oireyhtymätyypit
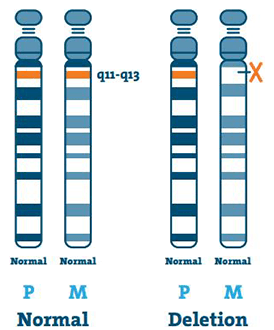
Poisto ( 65-75%)
DNA (deoksiribonukleiinihappo) on kromosomien pääkomponentti. Se sisältää ainutlaatuisen geneettisen koodimme. Suurimmasta osasta AS-potilaita puuttuu osa DNA: ta alueella 15q11-13 äidin kromosomissa 15.
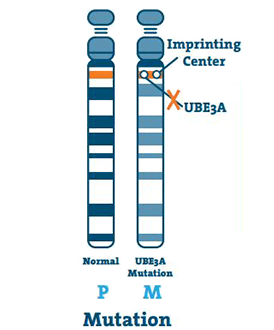
Mutaatio (5 -11%)
Tämä tapahtuu, kun UBE3A-geenin DNA: ssa on pieni poikkeama. Mutaatio voi tapahtua missä tahansa geenin kohdassa.
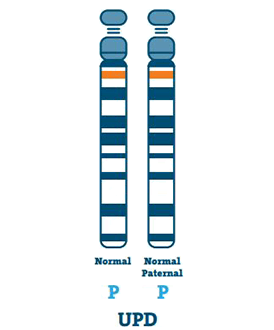
Yksisuuntainen parisairaus (3-7%)
UPD-potilaalla on kaksi kopiota isänsä kromosomista 15 isänsä ja äitinsä sijasta. UPD tapahtuu yleensä, jos munassa ei ole kromosomia 15.
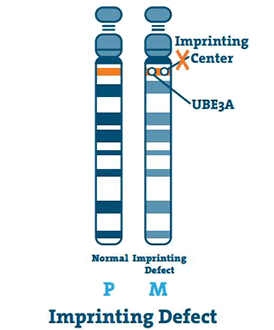
Tulostusvirhe (< 3%)
Tulostuskeskus on pieni DNA-jakso, joka sijaitsee kromosomin alueella q11-13. Harvinaisissa tapauksissa äidin kromosomi 15 on tyhjä, ja keskus kopioi isän kromosomin 15. Tämä on painovirhe.
Ominaisuudet
AS: n tyypilliset ominaisuudet eivät yleensä näy syntymä. Häiriöpotilailla on ruokinta-vaikeuksia imeväisikäisinä ja huomattava viivästynyt kehitys noin 6–12 kuukauden iässä. He tarvitsevat intensiivihoitoja toiminnallisten taitojen kehittämiseksi. AS vaikuttaa jokaiseen kilpailuun ja molempiin sukupuoliin. Se diagnosoidaan usein väärin autismiksi tai aivohalvaukseksi.
Käyttäytymispiirteet
Angelmanin oireyhtymää sairastavilla ihmisillä on joitain selkeitä käyttäytymispiirteitä, mukaan lukien onnellinen käytös, jolle on ominaista usein nauraminen, hymyily ja kiihottuvuus. Monet AS-potilaat ovat kiehtoneet vettä ja nauttivat suuresti uimasta ja uimisesta.
Angelmanin oireyhtymän syitä
Angelmanin oireyhtymä on yksittäisen geenin häiriö, jonka aiheuttaa menetys. toiminnasta UBE3A-geenissä äidin 15. kromosomissa. Ihmisillä on kaksi kromosomiryhmää – yksi peritty äidiltä ja toinen isältä. Tyypillisessä ihmisessä äidin perimä UBE3A-geeni on aktiivinen, kun taas isältä peritty geenikopio hiljennetään aivojemme hermosoluissa – ilmiö, joka tunnetaan nimellä painatus. AS-ihmisille tämä äidin geeni ei tee tehtäväänsä, ja se vaikuttaa heidän Messenger-RNA: han (mRNA).
![]()
Mikä on mRNA?
mRNA on kehon FedEx. DNA käyttää mRNA: ta toimituspalveluna piirustusten lähettämiseen solujemme proteiiniasennustehtaille. AS-potilailla on mutaatio, deleetio tai muu vika UBE3A-geenissään, joka keskeyttää tämän jakelupalvelun. Tämän seurauksena niiden neuronit eivät tuota mitään toiminnallista UBE3A-proteiinia, ja tämä laukaisee AS: n oireet. Tämä proteiini auttaa meitä kävelemään, puhumaan ja suorittamaan muita jokapäiväisiä tehtäviä.
![]()
Onko se geneettistä?
Useimmissa tapauksissa Angelmanin oireyhtymä ei ole perinnöllinen – etenkin poisto tai UPD. Sen sijaan nämä geneettiset muutokset tapahtuvat satunnaisina tapahtumina lisääntymissolujen muodostumisen tai alkion alkuvaiheen aikana.
Testit & Diagnoosi
Opi kriteereistä, joita lääkärit käyttävät selvittääkseen, onko henkilöllä Angelmanin oireyhtymä, ja lue käytettävissä olevista testeistä ja yleisistä virheellisistä diagnooseista.
![]()
Testaus
![]()
Diagnoosi

Liity yhteisöömme
Pidä yhteyttä muihin ihmisiin, joilla on lapsi tai rakkaansa, jolla on Angelmanin oireyhtymä.
