Einführung
Seit Jahrzehnten ist bekannt, dass Wechselwirkungen zwischen Nahrungsmitteln und Arzneimitteln (FDI) und Wechselwirkungen zwischen Kräutern und Arzneimitteln den Erfolg medizinischer Behandlungen einschränken. Die enorme Anzahl möglicher Wechselwirkungen zwischen genetischen Variationen, medizinischen Regimen und den zahlreichen bioaktiven Verbindungen in Lebensmitteln und Kräutern führt zu einer überwältigenden Komplexität. Moderne Tools wie Big-Data-Analyse, maschinelles Lernen und Simulation von Protein-Ligand-Wechselwirkungen können uns helfen, eine ganze Reihe von Fragen zu beantworten: Könnte die Auswahl von Lebensmitteln zum Scheitern therapeutischer Regime beitragen, und wenn ja, wie? Welche Lebensmittel sollten vor der Einnahme eines verschriebenen Arzneimittels konsumiert werden? Und wahrscheinlich die aufregendste Frage: Wie können wir diese Tools verwenden, um persönliche ausländische Direktinvestitionen vorherzusagen? Offensichtlich liegen viele Antworten im Metabolismus von Arzneimitteln, Nahrungsmitteln und Kräutern durch Cytochrom P450 3A4 (CYP3A4) in Leber und Verdauungstrakt (Galetin et al., 2010; Basheer und Kerem, 2015).
Die Mehrzahl der für CYP-Enzyme kodierenden Gene ist polymorph. Bisher ist das Pharmacogene Variation Consortium1, in dem weniger als 100 CYP3A4-Allele vertreten sind, die umfassendste Informationsquelle für CYP-Allele. Von diesen sind weniger als 40 exonische SNPs (Einzelnukleotidpolymorphismen), die zu einer modifizierten Proteinsequenz führen. Die geringe Anzahl von Probanden in allen zuvor veröffentlichten Arbeiten zu CYP3A4-Mutationen liefert uns begrenzte Daten zu den tatsächlichen Häufigkeiten von CYP3A4-Mutationen in der gesamten Population und in definierten Gruppen.
Nicht nur die zuverlässigen Informationen über die Inzidenz von SNPs sind unvollständig Auch ihre klinischen Auswirkungen sind in den meisten Fällen noch unklar (Zanger et al., 2014). Zu verstehen, welche und wann SNPs klinische Bedeutung haben könnten, ist eine äußerst komplexe Aufgabe. In-vitro-Tests sind zeitaufwändig, teuer und angesichts der großen Anzahl von Mutationen und der endlosen Anzahl von Lebensmittel-Arzneimittel-Kombinationen praktisch von geringer Relevanz. Molekulare Modellierungsmethoden, einschließlich Docking- und Freie-Energie-Bindungsberechnungen, können dazu dienen, mögliche Auswirkungen von SNPs und vieler Verbindungen auf den CYP3A4-vermittelten Metabolismus vorherzusagen (Lewis et al., 1998). Beispielsweise tragen nichtkovalente, hydrophobe, elektrostatische und Van-der-Waals-Wechselwirkungen zur Orientierung einer Verbindung und damit zu ihrer Bindung und Reaktion am aktiven Zentrum eines Enzyms bei. Diese bestimmen wiederum die Affinität und Spezifität des Enzyms zu verschiedenen Substraten und die Wirksamkeit von Enzyminhibitoren (Kirchmair et al., 2012; Basheer et al., 2017).
Hier schlagen wir eine neue vor Ansatz zur Messung der Allelfrequenz von CYP3A4-Mutationen in verschiedenen ethnischen Gruppen. Dieser umfassende Ansatz hat die Fähigkeit, Mutationen hervorzuheben, die in bestimmten ethnischen Gruppen vorherrschen, und in Kombination mit dem Screening auf wechselwirkende Chemikalien, z. B. Inhibitoren aus Lebensmitteln, die Aufklärung der Auswirkungen bestimmter Mutationen auf die Wechselwirkung zwischen Arzneimitteln und Lebensmitteln zu ermöglichen Schritt in Richtung personalisierte Medizin und Ernährung. Diese Arbeit kann das Bewusstsein für die mögliche klinische Bedeutung proteinverändernder CYP3A4-SNPs schärfen und schlägt auch einige notwendige Instrumente für die Förderung und Anwendung von Präzision und personalisierter Medizin vor.
Materialien und Methoden
Datenbank-Screening und Datenanalyse
Der Datensatz der CYP3A4-Varianten wurde vom gnomAD-Browser2 als CVS-Datei heruntergeladen. Python 2.7 mit NumPy-, Pandas- und Matplotlib-Paketen wurde zur Datenanalyse und -visualisierung verwendet (siehe ergänzendes Datenblatt S1). Agglomeratives hierarchisches Clustering wurde unter Verwendung der Expander 7-Software (Shamir et al., 2005) mit dem Pearson-Rangkorrelationskoeffizienten als Maß für Ähnlichkeiten und vollständigen Verknüpfungstyp durchgeführt. Für die Gruppierung von SNPs wurde ein Abstandsschwellenwert von 0,6 festgelegt.
In silico Polymorphism Modeling
wurde Maestro 2017-2 (Schrodinger, New York, NY, USA) für die Computermodellierung. Das CYP3A4-Docking-Modell wurde wie zuvor beschrieben gebaut (Basheer et al., 2017). Kurz gesagt, die CYP3A4-Kristallstruktur (PDB-Eintrag 2V0M) wurde gemäß den Schritten des Proteinvorbereitungs-Assistenten verarbeitet, modifiziert und verfeinert. Ein Docking-Gitter mit einer Metallkoordinationsbeschränkung für Fe2 + in der Hämgruppe wurde basierend auf dem Schwerpunkt von Ketoconazol an der ursprünglichen Bindungsstelle in der Kristallstruktur erzeugt. Sieben Mutationen wurden für Docking-Simulationen ausgewählt, eine als Vertreter für jede ethnische Gruppe (Tabellen 1, 2). Für jede Proteinvariante wurde vor den Proteinherstellungsschritten eine Einzelpunktmutation eingeführt. 3D-Strukturen von Liganden wurden basierend auf 2D-Strukturen von PubChem3 generiert und für das Andocken unter Verwendung der LigPrep-Aufgabe vorbereitet.Das OPLS3-Kraftfeld und die Standard-Gleitoptionen für die Standardgenauigkeit wurden für das Docking-Modell angewendet, mit der Ausnahme, dass die Metallkoordinationsbeschränkung verwendet wurde, sowie 30 Posen für die Anzahl der einzuschließenden Posen und 10 Posen für die Anzahl der zu schreibenden Posen aus. Für jeden Liganden wurde das Docking-Ergebnis mit dem niedrigsten Glide-Emodel-Score ausgewählt.

Tabelle 1. Ausgewählte repräsentative SNPs für sieben ethnische Gruppen.

Tabelle 2. Häufigkeit (%) ausgewählter Mutationen nach ethnischen Gruppen.
Ergebnisse
Die Genomaggregationsdatenbank (gnomAD; siehe Text Fußnote 2) aggregiert sowohl Exom- als auch Genomsequenzierungsdaten aus einer Vielzahl von großen Sequenzierungsprojekten. Es enthält Daten von 125.748 Exomsequenzen und 15.708 Gesamtgenomsequenzen von 141.456 nicht verwandten Personen, die sieben ethnische Populationen repräsentieren (Lek et al., 2016). Die GnomAD-Datenbank enthält 856 Varianten von CYP3A4, von denen 397 intronisch und 459 exonisch sind. Von den exonischen SNPs sind 312 Missense-Mutationen, was darauf hinweist, dass sie die Proteinstruktur beeinflussen. Das CYP3A4-Gen ist 34.205 bp lang. Seine 13 Exons umfassen eine 1.512-bp-kodierende Region, die ein Protein mit 504 Aminosäuren produziert. Die 412 exonischen SNPs mit einzigartigen Positionen in diesem Gen führen zu einer exonischen SNP-Dichte von 272 / kbp (Ergänzungstabelle S1). Die Berechnung der unterschiedlichen Allelfrequenzen pro ethnischer Gruppe zeigt, dass einige Populationen höhere Mutationshäufigkeiten aufweisen (Fig. 1A). Die meisten CYP3A4-Mutationen in der europäischen Bevölkerung sind in der Tat selten, wie allgemein angenommen wird, während Mutationen in anderen Populationen wie Afrika und Ostasien viel häufiger auftreten (Ergänzungstabelle S2).
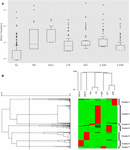
Abbildung 1. Analyse von CYP3A4-Missense-SNPs in sieben verschiedenen Populationen. (A) Log-Scale-Box-Plot der Allelfrequenzen. Kästchen repräsentieren den Interquartilbereich (IQR), blaue Linien repräsentieren den Median, Whisker repräsentieren Daten innerhalb von 1,5 IQR und Ausreißer werden als kleine Kreise angezeigt. (B) Hierarchische Clusterbildung von Allelfrequenzen. Jede Zeile repräsentiert einen einzelnen SNP. Jede Spalte repräsentiert eine unterschiedliche ethnische Bevölkerung. Die Allelfrequenz der SNPs in jeder der Populationen wird durch die Farbe der entsprechenden Zelle in der Matrixdatei dargestellt. Grün und Rot stehen für niedrige bzw. hohe Frequenzen. Das obere Dendrogramm zeigt Ähnlichkeiten im Allelfrequenzmuster zwischen jeder Gruppe von Probanden. Das linke Dendrogramm repräsentiert die Clusterbildung von Genen in zwei Gruppen. Die gestrichelte Linie stellt den Schwellenwert von 0,6 für die Aufteilung in Gruppen dar. EU – Europäer (nicht finnisch; n = 64.603), FIN – Europäer (finnisch; n = 12.562), ASH J – aschkenasischer Jude (n = 5.185), LTN – Latino (n = 17.720), AFR – Afrikaner (n =) 12.487), E ASN – Ostasien (n = 9.977), S ASN – Südasien (n = 64.603).
Wir haben hierarchisches Clustering verwendet, um Varianten mit ähnlichen Frequenzmustern zu gruppieren. Unsere Datenanalyse ergab sieben verschiedene Cluster (Abbildung 1B). Ferner wird deutlich beobachtet, dass hochfrequente SNPs in jedem Cluster für eine bestimmte Population charakteristisch sind. Die hierarchische Clusteranalyse der ethnischen Gruppen unterstützt den Zusammenhang zwischen genetischer Varianz und ethnischer Zugehörigkeit, indem verwandte Ethnien wie Süd- und Ostasiaten sowie finnische und nicht finnische Europäer zusammengefasst werden.
Zur Bewertung wurde ein Rechenmodell verwendet der mögliche Einfluss von Punktmutationen in CYP3A4 auf seine Fähigkeit, Substrate und Inhibitoren zu binden. CYP3A4 ist in der Lage, eine Vielzahl von endogenen und xenobiotischen Verbindungen zu oxidieren. Hier wurde Ketoconazol als repräsentatives Medikament und als sehr wirksamer spezifischer Inhibitor ausgewählt; Androstendion und Testosteron wurden als repräsentatives endogenes Hormon ausgewählt; und Demethoxycurcumin und Epigallocatechin wurden als Vertreter von diätetischen Bioaktivstoffen ausgewählt. Ein Docking-Modell wurde erstellt, um die Bindungspositionen der ausgewählten Verbindungen an der CYP3A4-Bindungsstelle vorherzusagen. Das Modell wurde zuerst validiert, indem die Ketoconazol-Pose an der Bindungsstelle mit einer RMSD von 1,52 Å relativ zur ursprünglichen Kristallstruktur erfolgreich wiederhergestellt wurde. Sieben mutierte Proteine wurden basierend auf der Kristallstruktur des Wildtyp-Proteins entworfen (ergänzende Abbildung S1). Für jede ethnische Gruppe wurde die häufigste eindeutige Mutation als repräsentativ ausgewählt. Die Wirkung einzelner Mutationen auf die Substratbindung wurde basierend auf dem Vergleich zwischen Docking-Posen auf dem nativen Protein und auf varianten Proteinen bewertet. Änderungen der Docking-Posen in Bezug auf RMSD sind in Tabelle 3 zusammengefasst.

Tabelle 3. RMSD relativ zur WT von Docking-Liganden an Bindungsstellen von sieben CYP3A4-Varianten.
Die Wirkung von CYP3A4-SNP auf die Substratbindung erwies sich als mutationssubstratspezifisch. Nur in wenigen Fällen verursachten Mutationen eine Änderung der Bindungsposition eines Liganden in der Bindungstasche. Die Testosteron-Docking-Pose war in allen sieben getesteten Varianten gleich. Die Varianten E262K, D174H und K168N verursachten in keinem der getesteten Moleküle eine Änderung der Bindungsposition. Die Mutationen L373F und T163A veränderten jedoch die Bindungsposition von Androstendion, so dass es parallel zur Hämgruppe und nicht senkrecht dazu positioniert war, wie im WT-Protein. Außerdem wurde Androstendion so gedreht, dass sich die Cyclopentanongruppe proximal zum Häm befindet, anstelle der Cyclohexanongruppe im WT-Protein. Die Mutationen S222P und L293P verursachten nur eine geringe Rotation in der Bindungsposition von Androstendion (Abbildung 2A). Von allen untersuchten Mutationen verursachte nur S222P wesentliche Veränderungen in den Docking-Posen von Ketoconazol und Demethoxycurcumin an der Bindungsstelle (2B, C); wohingegen für Epigallocatechin die Pose ändernde Mutation L373F war (2D).
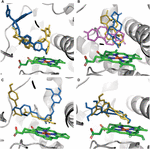
Abbildung 2. Modelle von Liganden, die an der Bindungsstelle von CYP3A4 angedockt sind. (A) Ketoconazol, (B) Androstendion, (C) Demethoxycurcumin und (D) Epigallocatechin. Die Proteinbindungsstelle wird durch graue Bänder dargestellt; Häm wird durch grüne Stäbchen dargestellt, Docking-Posen im WT-Protein und in S222P- und L373F-Mutanten sind sie als orange, blaue bzw. violette Stäbchen dargestellt. Androstendion-Docking-Posen in L293P- und T136A-Varianten überlappen die Posen in S222P- bzw. L373F-Varianten.
Diskussion
Cytochrom P450 3A4 ist das Hauptenzym, das für Wechselwirkungen zwischen Lebensmitteln und Arzneimitteln verantwortlich ist. Die aktuelle Forschung zu Mutationen in CYP3A4 konzentrierte sich auf einige Dutzend SNPs, die in bestimmten Studien gefunden wurden (Sata et al., 2000; Dai et al., 2001; Eiselt et al., 2001; Hsieh et al., 2001; Lamba et al ., 2002; Murayama et al., 2002). Wie hier gezeigt, stellen sie die Spitze eines Eisbergs dar, wenn man die Prävalenz und die möglichen Ergebnisse von CYP3A4-Mutationen berücksichtigt. Die Fülle an Projekten zur Sequenzierung großer Genome und Exome hat einen neuen Weg für die Identifizierung vieler unbekannter Mutationen eröffnet. Hier zeigen wir, dass die zuvor vorgestellten Mutationen nur die Spitze des Eisbergs sind, indem wir 856 in CYP3A4 vorhandene Mutationen demonstrieren, von denen ein Drittel die Proteinstruktur modifiziert. Unter Verwendung einer Kohorte von 141.456 nicht verwandten Personen wurde die genaue Allelfrequenz von CYP3A4-Mutationen für sieben verschiedene Ethnien berechnet. Nach unserem besten Wissen ist dies die größte und umfassendste Großdatenstudie zu exonischen CYP3A4-Mutationen und ihren Allelfrequenzen in verschiedenen Populationen, die bisher veröffentlicht wurde.
Polymorphe CYP3A4-Enzyme können für die Erklärung sehr wichtig sein Unterschiede in der Wirksamkeit und Toxizität des Arzneimittels zwischen verschiedenen Personen. Mutationen im CYP3A4-Gen können zu einer Aufhebung, Verringerung, Veränderung oder Erhöhung der enzymatischen Aktivität führen. Exonische Mutationen können die enzymatische Aktivität verändern, wie in einigen klinischen Studien mit ausgewählten Substraten gezeigt wurde. Einige Fälle von verändertem Metabolismus aufgrund von SNPs in CYP3A4 wurden bereits in der Literatur beschrieben (Eiselt et al., 2001; Miyazaki et al., 2008). Trotz der funktionellen Bedeutung und klinischen Relevanz von SNPs in CYP3A4 und möglicherweise aufgrund ihrer relativ geringen identifizierten Häufigkeit in der Allgemeinbevölkerung hat der Polymorphismus in CYP3A4 nicht die Aufmerksamkeit erhalten, die er verdient.
Hier dienten sieben Mutationen zur Vorhersage die Wirkung von SNPs auf die Substrat- und Inhibitor-Bindungsorientierung. In der Literatur unterteilt der CYP3A4-Polymorphismus die Allgemeinbevölkerung in drei Gruppen – schlechte Metabolisierer, normale Metabolisierer und schnelle Metabolisierer, basierend auf intronischen SNPs, die die Expressionsniveaus und nicht die Struktur modifizieren (Zanger und Schwab, 2013). Unsere Berechnungen legen eine zusätzliche Klassifizierung nahe: die veränderten Metabolisierer. Einige von unserem virtuellen Modell vorgeschlagene Mutationen würden eine Änderung der Bindungsorientierung einzelner Liganden verursachen. Es ist zu erwarten, dass diese Änderungen die Wahrscheinlichkeit einer enzymatischen Oxidation aufgrund eines größeren Abstands vom Häm verringern oder zu Produkten führen, die bei Toxizitätstests, die im Rahmen des Arzneimittelentwicklungsprozesses durchgeführt werden, ansonsten nicht erkennbar wären. Wie unser Modell vorhersagt, sind CYP3A4-Mutationen für die meisten Substrate gutartig.
Die modifizierte Position eines Substrats in der Bindungstasche aufgrund von Proteinstrukturänderungen ist nur ein möglicher Mechanismus, durch den eine Mutation die Aktivität eines Proteins verändern könnte . Eine beeinträchtigte Verankerung des Proteins an der Membran, beschädigte Substrat-führende Kanäle und ein beeinträchtigter Austritt der Produkte bieten zusätzliche Mechanismen für eine Mutationsänderung der Aktivität eines Proteins. Wie hier gezeigt, ist der Effekt jeder Mutation substratspezifisch.Die Bestimmung, welche Kombinationen von Substraten und Mutationen die enzymatische Aktivität mithilfe traditioneller In-vitro-Methoden verändern könnten, ist mühsam und unterstreicht die Notwendigkeit prädiktiver virtueller Werkzeuge zur Lösung dieses komplexen Rätsels.
Öffentliches und berufliches Interesse an persönlicher und Präzisionsmedizin wächst schnell. Die Vorhersage des modifizierten Arzneimittelstoffwechsels auf der Grundlage des individuellen Polymorphismus in CYP3A4 scheint nur eine Frage der Zeit zu sein. Hier schlagen wir vor, dass verschiedene ethnische Gruppen einzigartige Sätze von CYP3A4-SNPs tragen. In der Tat kann die ethnische Zugehörigkeit als erster machbarer Schritt in der personalisierten Medizin dienen, bevor ein individuelles DNA-Screening für alle durchgeführt wird. Interessanterweise hat die ethnische Zugehörigkeit eine weitere Auswirkung auf den CYP3A4-Arzneimittelstoffwechsel und ist ein wichtiger Faktor bei der Bestimmung der Ernährungsgewohnheiten und Ernährungsgewohnheiten. Es kann vorgeschlagen werden, Therapiesysteme speziell für jede ethnische Gruppe zu entwickeln, zumindest für Arzneimittel, die durch CYP3A4 stark metabolisiert werden. Dies unterstreicht die Möglichkeiten zur Nutzung und Integration von Datenbanken und zum vertieften Lernen, um festzustellen, wie SNPs, ethnische Zugehörigkeit, diätetische Verbindungen und Medikamente die CYP3A4-Aktivität und den Erfolg eines medizinischen Regimes verändern.
Datenverfügbarkeit
In dieser Studie wurden öffentlich verfügbare Datensätze analysiert. Diese Daten finden Sie hier: http://gnomad.broadinstitute.org/gene/ENSG00000160868.
Autorenbeiträge
Alle aufgeführten Autoren haben einen wesentlichen, direkten und intellektuellen Beitrag zum Arbeit und genehmigte es zur Veröffentlichung.
Interessenkonflikterklärung
Die Autoren erklären, dass die Forschung in Abwesenheit von kommerziellen oder finanziellen Beziehungen durchgeführt wurde, die als potenziell ausgelegt werden könnten Interessenkonflikt.
Ergänzungsmaterial
ABBILDUNG S1 | 3D-Bandmodell von CYP3A4 und die Position der mutierten Aminosäuren in den sieben zum Andocken entwickelten Proteinvarianten. Häm wird als grüne Stäbchen dargestellt, Fe2 + wird als rote Kugel dargestellt, SNPs, die in der In-Silico-Analyse verwendet werden, werden als rote Bereiche am Band dargestellt und R-Gruppen mutierter Aminosäuren in Variantenmodellen werden explizit als hellgraue Stäbchen dargestellt.
TABELLE S1 | CYP3A4-SNP-Typen in einer Bevölkerung von 141.456 nicht verwandten Personen, die 7 ethnische Populationen repräsentieren.
TABELLE S2 | CYP3A4-SNPs nach ethnischen Gruppen.
Fußnoten
- ^ www.pharmvar.org
- ^ https://gnomad.broadinstitute.org
- ^ https://pubchem.ncbi.nlm.nih.gov