소개
수십 년 동안 식품-약물 상호 작용 (FDI)과 약초-약물 상호 작용은 치료의 성공을 제한하는 것으로 알려져 왔습니다. 유전 적 변이, 의학적 정권, 식품과 허브에서 발견되는 수많은 생리 활성 화합물 사이의 가능한 상호 작용이 엄청나게 많기 때문에 엄청난 복잡성이 발생합니다. 빅 데이터 분석, 기계 학습, 단백질-리간드 상호 작용 시뮬레이션과 같은 최신 도구는 전체 질문에 답하는 데 도움이 될 수 있습니다. 음식 선택이 치료 요법의 실패에 기여할 수 있으며, 그렇다면 어떻게할까요? 처방약을 복용하기 전에 어떤 음식을 섭취해야합니까? 그리고 아마도 가장 흥미로운 질문 일 것입니다. 개인 FDI를 예측하기 위해 이러한 도구를 어떻게 사용할 수 있습니까? 분명히 많은 답변이 간과 소화관에서 사이토 크롬 P450 3A4 (CYP3A4)에 의한 약물, 음식 및 허브의 대사에 있습니다 (Galetin et al., 2010; Basheer and Kerem, 2015).
CYP 효소를 암호화하는 유전자의 대부분은 다형성입니다. 현재까지 CYP 대립 유전자를 자세히 설명하는 가장 포괄적 인 정보 소스는 CYP3A4의 대립 유전자가 100 개 미만인 Pharmacogene Variation Consortium1입니다. 이 중 40 개 미만이 변형 된 단백질 서열을 생성하는 엑 소닉 SNP (단일 뉴클레오티드 다형성)입니다. CYP3A4 돌연변이에 대한 이전에 발표 된 모든 연구에서 소수의 피험자는 전체 모집단과 정의 된 그룹에서 CYP3A4 돌연변이의 실제 빈도에 관한 제한된 데이터를 제공합니다.
SNP 발생률에 대한 신뢰할 수있는 정보가 불완전 할뿐만 아니라 , 또한 대부분의 경우 임상 적 의미가 아직 명확하지 않습니다 (Zanger et al., 2014). SNP가 임상 적 중요성을 가질 수있는시기와 SNP를 이해하는 것은 매우 복잡한 작업입니다. 체외 분석은 많은 양의 돌연변이와 끝없는 수의 식품-약물 조합을 고려할 때 시간이 많이 걸리고 비용이 많이 들고 실질적으로 관련성이 낮습니다. 도킹 및 자유 에너지 결합 계산을 포함한 분자 모델링 방법은 CYP3A4 매개 대사에 대한 SNP 및 많은 화합물의 잠재적 효과를 예측하는 데 도움이 될 수 있습니다 (Lewis et al., 1998). 예를 들어, 비공유, 소수성, 정전기 및 van der Waals 상호 작용은 모두 화합물의 방향에 기여하므로 효소의 활성 부위에서 결합 및 반응합니다. 차례로, 이들은 다른 기질에 대한 효소의 친 화성 및 특이성 및 효소 억제제의 효능을 결정합니다 (Kirchmair et al., 2012; Basheer et al., 2017).
여기에서 새로운 것을 제안합니다. 다른 인종 그룹에서 CYP3A4 돌연변이의 대립 유전자 빈도를 측정하는 방법. 이 포괄적 인 접근 방식은 특정 인종 그룹에서 만연한 돌연변이를 강조 할 수있는 힘을 가지고 있으며, 상호 작용하는 화학 물질에 대한 스크리닝과 결합됩니다. 예를 들어 식품의 억제제는 약물-식품 상호 작용에 대한 특정 돌연변이의 효과를 설명 할 수 있습니다. 맞춤형 의약품 및 영양을 향한 발걸음. 이 연구는 단백질을 변경하는 CYP3A4 SNP의 임상 적 중요성에 대한 인식을 높이고 정밀 및 맞춤 의학의 홍보 및 적용에 필요한 몇 가지 도구를 제안합니다.
재료 및 방법
데이터베이스 스크리닝 및 데이터 분석
CYP3A4 변형 데이터 세트는 gnomAD browser2에서 CVS 파일로 다운로드되었습니다. NumPy, pandas 및 matplotlib 패키지가 포함 된 Python 2.7이 데이터 분석 및 시각화에 사용되었습니다 (보충 데이터 시트 S1 참조). Expander 7 소프트웨어 (Shamir et al., 2005)를 사용하여 유사성 및 완전한 연결 유형의 척도로 Pearson 순위 상관 계수를 사용하여 집계 계층 적 클러스터링을 수행했습니다. SNP 그룹화를 위해 거리 임계 값 0.6이 설정되었습니다.
In silico Polymorphism Modeling
Maestro 2017-2 릴리스 (Schrodinger, New York, NY, United States)는 계산 모델링. CYP3A4 도킹 모델은 이전에 설명한대로 구축되었습니다 (Basheer et al., 2017). 간단히 말해서, CYP3A4 결정 구조 (PDB 항목 2V0M)는 단백질 준비 마법사 단계에 따라 처리, 수정 및 정제되었습니다. 헴 그룹의 Fe2 +에 대한 금속 배위 제약을 갖는 도킹 그리드는 결정 구조의 원래 결합 부위에있는 케토코나졸의 중심을 기반으로 생성되었습니다. 도킹 시뮬레이션을 위해 7 개의 돌연변이가 선택되었으며, 하나는 각 인종 그룹을 대표합니다 (표 1, 2). 각 변이 단백질에 대해 단백질 준비 단계 전에 단일 점 돌연변이가 도입되었습니다. PubChem3의 2D 구조를 기반으로 리간드의 3D 구조를 생성하고 LigPrep 작업을 사용하여 도킹을 준비했습니다.표준 정밀도에 대한 OPLS3 포스 필드 및 기본 글라이드 옵션이 도킹 모델에 적용되었습니다. 단, 금속 조정 제약 조건이 사용 된 경우와 포함 할 포즈 수에 대해 30 개 포즈, 작성할 포즈 수에 대해 10 개 포즈가 사용되었습니다. 밖. 각 리간드에 대해 Glide emodel 점수가 가장 낮은 도킹 결과가 선택되었습니다.

표 1. 7 개 민족 그룹에 대해 선택된 대표 SNP.

표 2. 인종 그룹 별 선택된 돌연변이의 빈도 (%).
결과
Genome Aggregation Database (gnomAD, 텍스트 각주 2 참조)는 다양한 대규모 시퀀싱 프로젝트의 엑솜 및 게놈 시퀀싱 데이터를 모두 집계합니다. 여기에는 7 개의 인종 집단을 대표하는 141,456 명의 관련되지 않은 개인의 125,748 개의 엑솜 서열과 15,708 개의 전체 게놈 서열의 데이터가 포함됩니다 (Lek et al., 2016). GnomAD 데이터베이스는 CYP3A4의 856 개의 변종을 제공하며, 그중 397 개는 인트로 닉이고 최대 459 개는 엑 소닉입니다. 엑 소닉 SNP 중 312 개는 미스 센스 돌연변이로 단백질 구조에 영향을 미친다는 것을 나타냅니다. CYP3A4 유전자의 길이는 34,205bp입니다. 그것의 13 개의 엑손은 504 개의 아미노산으로 구성된 단백질을 생산하는 1,512-bp 코딩 영역으로 구성됩니다. 이 유전자에서 고유 한 위치를 가진 412 개의 엑 소닉 SNP는 272 / kbp의 엑 소닉 SNP 밀도를 나타냅니다 (보충 표 S1).
종족 별 대립 유전자 빈도를 계산하면 일부 집단이 더 높은 돌연변이 빈도를 나타냄을 알 수 있습니다. (그림 1A). 유럽 인구의 대부분의 CYP3A4 돌연변이는 일반적으로 생각하는 것처럼 실제로 드물지만 아프리카 및 동아시아와 같은 다른 인구의 돌연변이가 훨씬 더 널리 퍼져 있습니다 (보충 표 S2).
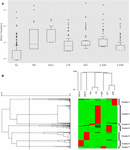
그림 1. 7 개의 개별 집단에서 CYP3A4 미스 센스 SNP 분석. (A) 대립 유전자 주파수의 로그 스케일 박스 플롯. 상자는 사 분위수 범위 (IQR)를 나타내고 파란색 선은 중앙값을 나타내고 수염은 1.5 IQR 내의 데이터를 나타내며 이상 값은 작은 원으로 표시됩니다. (B) 대립 유전자 빈도의 계층 적 클러스터링. 각 행은 단일 SNP를 나타냅니다. 각 열은 고유 한 민족 인구를 나타냅니다. 각 집단에서 SNP의 대립 유전자 빈도는 매트릭스 파일에서 해당 세포의 색상으로 표시됩니다. 녹색과 빨간색은 각각 저주파와 고주파를 나타냅니다. 상부 덴드로 그램은 각 피험자 그룹 간의 대립 유전자 빈도 패턴의 유사성을 보여줍니다. 왼쪽 덴드로 그램은 두 그룹의 유전자 군집을 나타냅니다. 파선은 그룹으로 분할하는 데 사용되는 0.6 거리 임계 값을 나타냅니다. EU – 유럽 (비 핀란드, n = 64,603), FIN – 유럽 (핀란드, n = 12,562), ASH J – Ashkenazi 유태인 (n = 5,185), LTN – 라틴계 (n = 17,720), AFR – 아프리카 (n = 12,487), E ASN – 동아시아 (n = 9,977), S ASN – 남아시아 (n = 64,603).
유사한 주파수 패턴을 가진 변형을 그룹화하기 위해 계층 적 클러스터링을 사용했습니다. 우리의 데이터 분석은 7 개의 별개의 클러스터를 산출했습니다 (그림 1B). 또한 각 클러스터의 고주파 SNP가 하나의 특정 인구에 특징적이라는 것이 명확하게 관찰되었습니다. 인종 그룹의 계층 적 군집 분석은 핀란드 및 비 핀란드 유럽인뿐만 아니라 남아시아 인과 동아시아 인과 같은 관련 민족을 함께 그룹화하여 유전 적 분산과 민족 간의 연관성을 지원합니다.
평가에는 계산 모델이 사용되었습니다. CYP3A4의 점 돌연변이가 기질과 억제제에 결합하는 능력에 미칠 수있는 영향. CYP3A4는 광범위한 내인성 및 이종 생물 화합물을 산화시킬 수 있습니다. 여기에서 케토코나졸은 대표적인 약물이자 매우 효율적인 특정 억제제로 선정되었습니다. 안드로 스텐 디온과 테스토스테론이 대표적인 내인성 호르몬으로 선정되었습니다. 그리고 demethoxycurcumin과 epigallocatechin은식이 생리 활성 물질의 대표자로 선정되었습니다. 도킹 모델은 CYP3A4 결합 부위에서 선택된 화합물의 결합 자세를 예측하기 위해 구축되었습니다. 이 모델은 원래 결정 구조에 비해 RMSD가 1.52Å 인 결합 부위에서 케토코나졸 포즈를 성공적으로 복원하여 처음 검증되었습니다. 야생형 단백질의 결정 구조를 기반으로 7 개의 돌연변이 단백질이 설계되었습니다 (보충 그림 S1). 각 인종 그룹에 대해 가장 빈번한 고유 돌연변이가 대표로 선택되었습니다. 기질 결합에 대한 단일 돌연변이의 효과는 고유 단백질과 변이체 단백질에 대한 도킹 포즈 간의 비교를 기반으로 평가되었습니다. RMSD 측면에서 도킹 포즈의 변경 사항은 표 3에 요약되어 있습니다.

표 3. 7 개의 CYP3A4 변이체의 결합 부위에 대한 도킹 리간드의 WT에 대한 RMSD.
기질 결합에 대한 CYP3A4 SNP의 효과는 돌연변이-기질 특이적인 것으로 밝혀졌습니다. 소수의 경우에만 돌연변이로 인해 결합 주머니에서 리간드의 결합 자세가 변경되었습니다. 테스토스테론 도킹 자세는 테스트 된 7 가지 변종 모두에서 동일했습니다. E262K, D174H 및 K168N 변이체는 테스트 된 분자에서 결합 포즈 변화를 일으키지 않았습니다. 그러나 L373F 및 T163A 돌연변이는 안드로 스텐 디온의 결합 포즈를 변경하여 WT 단백질에서와 같이 헴 그룹에 수직이 아닌 헴 그룹에 평행하게 위치하도록했습니다. 또한, 안드로 스텐 디온은 WT 단백질에서 시클로 헥사 논 그룹 대신에 시클로 펜타 논 그룹이 헴에 근접하도록 회전되었다. S222P 및 L293P 돌연변이는 안드로 스텐 디온의 결합 포즈에서 작은 회전만을 유발했습니다 (그림 2A). 조사 된 모든 돌연변이 중에서, S222P만이 결합 부위에서 케토코나졸 및 데메 톡시 쿠르쿠민의 도킹 포즈에 상당한 변화를 일으켰습니다 (그림 2B, C). 에피 갈로 카테킨의 경우 포즈 변경 돌연변이는 L373F였습니다 (그림 2D).
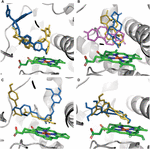
그림 2. CYP3A4의 결합 부위에 도킹 된 리간드 모델. (A) 케토코나졸, (B) 안드로 스텐 디온, (C) 데메 톡시 커큐민 및 (D) 에피 갈로 카테킨. 단백질 결합 부위는 회색 리본으로 표시됩니다. heme은 녹색 스틱으로 표시되며 WT 단백질 및 S222P 및 L373F 돌연변이 체의 도킹 포즈는 각각 주황색, 파란색 및 보라색 스틱으로 표시됩니다. L293P 및 T136A 변형의 Androstenedione 도킹 포즈는 각각 S222P 및 L373F 변형의 포즈와 겹칩니다.
토론
시토크롬 P450 3A4는 식품-약물 상호 작용을 담당하는 주요 효소입니다. CYP3A4의 돌연변이에 대한 현재 연구는 지정된 연구에서 발견 된 수십 개의 SNP에 초점을 맞추고 있습니다 (Sata et al., 2000; Dai et al., 2001; Eiselt et al., 2001; Hsieh et al., 2001; Lamba et al. ., 2002; Murayama et al., 2002). 여기에서 입증 된 바와 같이, 그들은 CYP3A4 돌연변이의 유병률과 잠재적 결과를 고려하여 빙산의 일각을 나타냅니다. 풍부한 대규모 게놈 및 엑솜 시퀀싱 프로젝트는 많은 알려지지 않은 돌연변이를 식별하는 새로운 길을 열었습니다. 여기에서, 우리는 이전에 제시된 돌연변이가 CYP3A4에 존재하는 856 개의 돌연변이를 보여줌으로써 빙산의 일각에 불과하다는 것을 보여 주며, 그중 1/3은 단백질 구조를 수정합니다. 관련되지 않은 141,456 명의 개인 코호트를 사용하여 CYP3A4 돌연변이의 정확한 대립 유전자 빈도가 7 개의 개별 민족에 대해 계산되었습니다. 우리가 아는 한, 이것은 현재까지 발표 된 다양한 집단의 CYP3A4 엑손 돌연변이 및 대립 유전자 빈도에 대한 가장 크고 포괄적 인 대규모 데이터 연구입니다.
다형성 CYP3A4 효소는 설명에 매우 중요 할 수 있습니다. 약물 효능과 독성의 차이. CYP3A4 유전자의 돌연변이는 효소 활성을 폐지, 감소, 변경 또는 증가시킬 수 있습니다. 엑손 돌연변이는 선택된 기질에 대한 몇 가지 임상 연구에서 입증 된 바와 같이 효소 활성을 수정할 수 있습니다. CYP3A4에서 SNP로 인한 대사 변화의 일부 사례는 이미 문헌에 설명되어 있습니다 (Eiselt et al., 2001; Miyazaki et al., 2008). CYP3A4에서 SNP의 기능적 중요성과 임상 적 관련성에도 불구하고 일반 인구에서 상대적으로 식별 된 빈도가 낮기 때문에 CYP3A4의 다형성은 주목할만한 관심을받지 못했습니다.
여기에서 7 개의 돌연변이가 예측에 기여했습니다. 기질 및 억제제 결합 방향에 대한 SNP의 효과. 문헌에서 CYP3A4 다형성은 구조보다는 발현 수준을 수정하는 인트로 닉 SNP를 기반으로 일반 인구를 열악한 대 사자, 정상 대 사자, 빠른 대 사자 세 그룹으로 나눕니다 (Zanger and Schwab, 2013). 우리의 계산은 추가 분류를 제안합니다 : 변경된 대사 물질. 가상 모델에 의해 제안 된 일부 돌연변이는 개별 리간드의 결합 방향을 변경합니다. 이러한 변화는 헴으로부터의 거리 증가로 인해 효소 적 산화 가능성을 감소 시키거나 약물 개발 과정의 일부로 수행되는 독성 테스트 동안 분명하지 않은 제품으로 이어질 것으로 예상됩니다. 그러나 우리 모델이 예측 한 바와 같이 대부분의 기질에서 CYP3A4 돌연변이는 양성입니다.
단백질 구조 변화로 인해 결합 주머니에서 기질의 위치가 수정 된 것은 돌연변이가 단백질의 활성을 바꿀 수있는 하나의 가능한 메커니즘 일뿐입니다. . 막에 대한 단백질 고정 장애, 기질 선도 채널 손상 및 제품의 손상된 출구는 단백질 활동의 돌연변이 변화에 대한 추가 메커니즘을 제공합니다. 여기에 표시된 것처럼 모든 돌연변이의 효과는 기질에 따라 다릅니다.기존의 시험관 내 방법을 사용하여 효소 활동을 수정할 수있는 기질과 돌연변이의 조합을 결정하는 것은 힘들며,이 복잡한 퍼즐을 해결하는 데있어 예측 가상 도구의 필요성을 강조합니다.
개인 및 정밀 의학에 대한 공공 및 전문적 관심 빠르게 성장하고 있습니다. CYP3A4의 개별 다형성을 기반으로 한 변형 약물 대사의 예측은 시간 문제인 것 같습니다. 여기, 우리는 고유 한 인종 그룹이 CYP3A4 SNP의 고유 한 세트를 품고 있다고 제안합니다. 실제로 민족성은 개인화 된 의료의 첫 번째 실행 가능한 단계로, 모두를위한 개별 DNA 스크리닝을 구현하기 전에 수행 할 수 있습니다. 흥미롭게도, 민족성은 CYP3A4 약물 대사에 대한 또 다른 의미를 가지고 있으며, 이는 음식 선택과 식습관을 결정하는 주요 요소입니다. 치료 요법은 적어도 CYP3A4에 의해 고도로 대사되는 약물에 대해 각 인종 그룹에 대해 특별히 설계되어야한다고 제안 될 수 있습니다. 이는 SNP, 민족성,식이 화합물 및 약물이 CYP3A4 활동과 의료 체제의 성공을 어떻게 수정하는지 식별하기 위해 데이터베이스와 딥 러닝을 활용하고 통합 할 수있는 기회를 강조합니다.
데이터 가용성
이 연구에서는 공개적으로 사용 가능한 데이터 세트를 분석했습니다. 이 데이터는 여기에서 찾을 수 있습니다. http://gnomad.broadinstitute.org/gene/ENSG00000160868.
저자 기고
나열된 모든 저자는 다음 사이트에 실질적이고 직접적이며 지적 기여를했습니다. 출판을 위해 승인했습니다.
이해 상충 선언문
저자는 연구가 잠재력으로 해석 될 수있는 상업적 또는 재정적 관계가없는 상태에서 수행되었다고 선언합니다. 이해 상충.
보충 자료
그림 S1 | CYP3A4의 3D 리본 모델 및 도킹 용으로 설계된 7 개의 변이 단백질에서 돌연변이 된 아미노산의 위치. Heme은 녹색 막대로, Fe2 +는 빨간색 구로, 인실 리코 분석에 사용 된 SNP는 리본의 빨간색 영역으로, 변형 모델에서 돌연변이 된 아미노산의 R 그룹은 밝은 회색 막대로 명시 적으로 표시됩니다.
표 S1 | CYP3A4 SNP 유형은 141 명, 7 개 민족 인구를 대표하는 456 명의 관련이없는 개인입니다.
표 S2 | 민족 별 CYP3A4 SNP
각주
- ^ www.pharmvar.org
- ^ https://gnomad.broadinstitute.org
- ^ https://pubchem.ncbi.nlm.nih.gov