Angelman syndrom (AS) er en sjelden neurogenetisk lidelse som rammer omtrent en av 15.000 mennesker – omtrent 500.000 individer over hele verden. Barn og voksne med AS har vanligvis balanseproblemer, nedsatt motorikk og svekkende anfall. Noen individer går aldri. De fleste snakker ikke. Forstyrrede søvnsykluser kan også være en alvorlig utfordring for den enkelte og vaktmester. Personer med AS trenger kontinuerlig omsorg og er ikke i stand til å leve selvstendig. De har en normal forventet levealder. Dette er livet i dag for mennesker som lever med Angelman syndrom, men håpet er her. Forskere mener at AS har størst potensial for å bli kurert sammenlignet med andre neurogenetiske lidelser, og FAST (Foundation for Angelman Syndrome Therapeutics) har en plan godt i gang for å oppnå nettopp det.
Typer Årsaker Tester & Diagnoseresurser
Typer av Angelman-syndromet
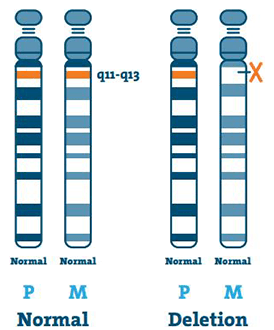
Sletting ( 65-75%)
DNA (deoksyribonukleinsyre) er hovedkomponenten i kromosomer. Den inneholder vår unike genetiske kode. De fleste individer med AS mangler et stykke DNA i region 15q11-13 på mors kromosom 15.
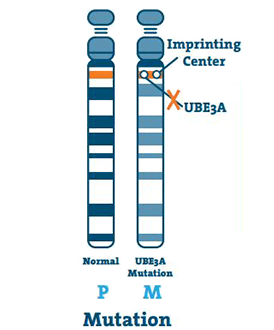
Mutasjon (5 -11%)
Dette skjer når det er en liten abnormitet i DNA til UBE3A-genet. En mutasjon kan skje hvor som helst på genet.
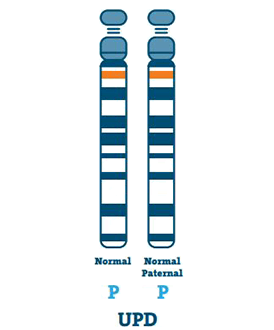
Uniparental Disomy (3-7%)
Et individ med UPD har to eksemplarer av kromosom 15 fra faren sin, i stedet for en hver fra faren og moren. UPD skjer vanligvis hvis det ikke er noe kromosom 15 i egget.
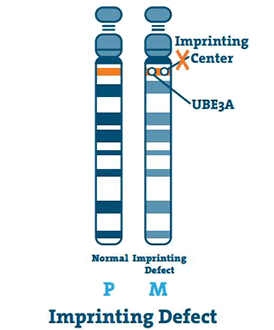
Imprinting Defect (< 3%)
Påtrykkingssenteret er en liten strekning av DNA som ligger i kromosomområdet q11-13. I sjeldne tilfeller er mors kromosom 15 tomt, og senteret kopierer farens kromosom 15. Dette er en pregefeil.
Egenskaper
Typiske kjennetegn ved AS er vanligvis ikke tydelige ved fødsel. Personer med sykdommen har fôringsvansker som spedbarn og merkbar forsinket utvikling rundt 6-12 måneder. De trenger intensive terapier for å utvikle funksjonelle ferdigheter. AS påvirker hvert løp og begge kjønn. Det blir ofte feildiagnostisert som autisme eller cerebral parese.
Behavioral Traits
Personer med Angelman syndrom har noen distinkte atferdstrekk, inkludert en lykkelig oppførsel, preget av hyppig latter, smilende og spennende. Mange individer med AS har en fascinasjon av vann og gleder seg stort over aktiviteter som bading og bading.
Årsaker til Angelmans syndrom
Angelman syndrom er en enkelt-gen lidelse forårsaket av tap av funksjon i UBE3A-genet på moderens 15. kromosom. Mennesker har to sett med kromosomer – en arvet fra moren og en fra faren. I en typisk person er det maternelt nedarvede UBE3A-genet aktivt, mens kopien av genet arvet fra faren blir tauset i nevronene i hjernen vår – et fenomen kjent som innprenting. For mennesker med AS gjør ikke dette modergenet jobben sin, og det påvirker Messenger-RNA (mRNA).
![]()
Hva er mRNA?
mRNA er kroppens FedEx. Vårt DNA bruker mRNA som en leveringstjeneste for å sende tegninger til proteinmonteringsfabrikkene til cellene våre. Personer med AS har en mutasjon, sletting eller annen defekt i UBE3A-genet som forstyrrer denne leveringstjenesten. Som et resultat lager ikke nevronene deres noe funksjonelt UBE3A-protein, og det er det som utløser symptomene på AS. Dette proteinet er det som hjelper oss å gå, snakke og utføre andre hverdagsoppgaver.
![]()
Er det genetisk?
I de fleste tilfeller arves ikke Angelman syndrom – spesielt de som er forårsaket av en sletting eller UPD. I stedet oppstår disse genetiske endringene som tilfeldige hendelser under dannelsen av reproduktive celler eller i tidlig embryonal utvikling.
Tester & Diagnose
Lær om kriteriene leger bruker for å avgjøre om et individ har Angelman-syndrom, og lese om tilgjengelige tester og vanlige feildiagnoser.
![]()
Testing
![]()
Diagnose

Bli med i samfunnet vårt
Få kontakt med andre mennesker som har et barn eller en kjær med Angelman-syndrom.
