Malign intravaskulær betennelse
Sepsis har blitt referert til som en prosess med ondartet intravaskulær betennelse . Normalt sikrer en kraftig, kompleks, immunologisk kaskade en hurtig beskyttende respons på mikroorganismens invasjon hos mennesker. Et mangelfullt immunologisk forsvar kan tillate at infeksjon blir etablert; imidlertid kan en overdreven eller dårlig regulert respons skade verten gjennom maladaptiv frigjøring av urfolksgenererte inflammatoriske forbindelser (se bildet nedenfor).
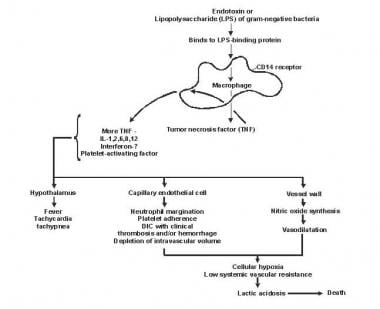 Patogenese av sepsis og multiorgansvikt.
Patogenese av sepsis og multiorgansvikt. Lipid A og andre bakterielle produkter frigjør cytokiner og andre immunmodulatorer som formidler de kliniske manifestasjonene av sepsis. Interleukiner, tumornekrosefaktor (TNF) -α, interferon gamma (IFN-γ) og andre kolonistimulerende faktorer produseres raskt i løpet av minutter eller timer etter interaksjoner av monocytter og makrofager med lipid A.
Inflammatorisk mediatorfrigjøring blir en selvstimulerende prosess, og frigjøring av andre slike mediatorer, inkludert interleukin (IL) -1, blodplateaktiverende faktor, IL-2, IL-6, IL-8, IL-10 og nitrogenoksid (NO), øker cytokinnivået ytterligere. Dette fører til fortsatt aktivering av polymorfonukleære leukocytter (PMN), makrofager og lymfocytter; proinflammatoriske meglere rekrutterer flere av disse cellene. Alle disse prosessene skaper en tilstand av destruktiv immunologisk dissonans.
Sepsis er beskrevet som en autodestruktiv prosess som tillater utvidelse av normal patofysiologisk respons på infeksjon for å involvere ellers normalt vev og resultater i MODS. Organdysfunksjon eller organsvikt kan være det første kliniske tegn på sepsis, og ingen organsystemer er immun mot konsekvensene av den inflammatoriske overdreven sepsis. Dødeligheten øker når organsvikt øker.
Selv om ukontrollert, når MODS utvikler systemisk bevis på både proinflammatorisk og antiinflammatorisk oppregulering, er vanligvis tilstede, noe som tyder på at svikt i vertsforsvarshomeostase er den siste veien fra sepsis til MODS, snarere enn enkel hypotensjon-indusert end-organskade, slik det kan oppstå med hemorragisk sjokk. Overlevelse fra alvorlig sepsis med MODS er vanligvis assosiert med en generalisert reduksjon i både proinflammatorisk og antiinflammatorisk respons.
En ny hypotese har nylig dukket opp at overlevelse fra alvorlig sepsis krever en generalisert nedregulering av kroppens immunrespons, energiske funksjoner og tilhørende organytelse. Dermed kan MODS ved vertsens adaptive respons på overveldende betennelse, slik at betennelse tømmes uten å forårsake permanent skade på enden. Som diskutert nedenfor, avslører alle organer en generalisert hyporesponsivitet som helt klart er unormal i helsen, men som kan markere en overlevelsesstrategi ved alvorlig sepsis.
Dysfunksjon i organsystemer
Sirkulasjonsforstyrrelse
Betydelig forstyrrelse i autoregulering av sirkulasjon er typisk for sepsis. Vasoaktive meglere forårsaker vasodilatasjon og øker mikrovaskulær permeabilitet på infeksjonsstedet. NO spiller en sentral rolle i vasodilatasjonen av septisk sjokk. Også nedsatt utskillelse av vasopressin kan forekomme, noe som kan tillate vedvarende vasodilatasjon.
Endringer i både systolisk og diastolisk ventrikulær ytelse forekommer i sepsis. Gjennom bruk av Frank-Starling-mekanismen øker hjertevolumet ofte for å opprettholde blodtrykket i nærvær av systemisk vasodilatasjon. Pasienter med allerede eksisterende hjertesykdom klarer ikke å øke hjertevolumet på riktig måte.
Regionalt forstyrrer sepsis den normale fordelingen av systemisk blodstrøm til organsystemer. Følgelig kan kjerneorganer ikke motta passende oksygenlevering, og resultatet er det som er kjent som regional hypoperfusjon.
Mikrosirkulasjon er det viktigste målorganet for skade ved sepsis siden vaskulært endotel er universelt påvirket av de sirkulerende inflammatoriske meglerne. Selv om det er uklart om mikrosirkulasjonsforstyrrelser er årsaken eller en uskyldig tilskuer til end organskaden, ses tydelig mikrovaskulær dysfunksjon. En reduksjon i antall perfuserte kapillærer er sett, selv om det ved bruk av vasodilatatorapi forekommer full mikrovaskulær rekruttering. Mitokondriell dysfunksjon forekommer også og er ofte assosiert med reduserte mitokondrie transmembrane potensielle gradienter, som er nødvendige for å drive oksidativ fosforylering. Sluttresultatet er en tilsynelatende manglende evne til å slutte organer til å trekke ut oksygen maksimalt.
Debatten fortsetter om denne svikten i energimetabolismen er en adaptiv cytobeskyttende mekanisme som ligner på dvalemodus eller gjenspeiler primær mitokondriepatologi. Dette er områder med aktiv forskning, men oversettes for tiden ikke til klare retningslinjer for klinisk praksis. Økt kapillær endotelpermeabilitet fører til utbredt proteinrikt vevsødem.
Septisk sjokk og SIRS er preget av reversibel hjerteinfarktdepresjon, som kan vise seg å være motstandsdyktig mot katekolamin og væskeadministrasjon. Sirkulerende «myocardial depressant factor» – som sannsynligvis representerer de synergistiske effektene av TNF-α, IL-1β, andre cytokiner og NO – er involvert i patogenesen. De to egenskapene til denne akutte stressmyokarddepresjonen er nedsatt adrenerg respons og diastolisk dysfunksjon som fører til relativ katekolaminresistens og små snarere enn utvidede hjerter Makrovaskulær myokardisk iskemi og hypoperfusjon er usannsynlig bidragsytere.
Ved alvorlig sepsis og septisk sjokk forårsaker mikrosirkulasjonsdysfunksjon og mitokondriell depresjon regional vevsnød, og regional dysoksi vedvarer derfor. Denne tilstanden kalles mikrosirkulasjons- og mitokondrie-nødsyndrom (MMDS). Sepsis-indusert inflammatorisk autoregulatorisk dysfunksjon vedvarer, og oksygenbehovet samsvares ikke med tilførsel, noe som fører til MODS.
Omfordeling av intravaskulært væskevolum som følge av redusert arteriell vaskulær tone, redusert venøs retur fra venøs di lasjon og frigjøring av myokardiale stoffer forårsaker hypotensjon.
Pulmonal dysfunksjon
Endotelskade i lungevaskulaturen fører til forstyrret kapillær blodstrøm og forbedret mikrovaskulær permeabilitet, noe som resulterer i interstitielt og alveolært ødem. Neutrofil inneslutning i lunges mikrosirkulasjonen initierer og forsterker skaden på alveolære kapillærmembraner. Akutt lungeskade og akutt respiratorisk nødsyndrom (ARDS) er hyppige manifestasjoner av disse effektene. Sepsis og lungebetennelse er faktisk de vanligste årsakene til ARDS.
Gastrointestinal dysfunksjon
Mage-tarmkanalen (GI) kan bidra til å forplante skaden av sepsis. Gjengroing av bakterier i øvre mage-tarmkanalen kan suges inn i lungene, noe som gir nosokomial eller aspirasjons lungebetennelse. Den normale barrierefunksjonen i tarmen kan påvirkes, slik at translokasjon av bakterier, endotoksiner og normale fordøyelsesproteaser tillates i systemisk sirkulasjon og utvider septisk respons.
Septisk sjokk kan forårsake paralytisk ileus som kan føre til en forsinkelse i institusjonen for enteral fôring. Overdreven NO-produksjon antas å være det forårsakende middel til sepsis-indusert ileus. Det optimale nivået av ernæringsinntak forstyrres med høye protein- og kaloribehov. Narkotika og muskelavslappende midler kan forverre MI-kanalens motilitet.
Leverdysfunksjon
Som en konsekvens av rollen leveren spiller i vertsforsvar kan de unormale syntetiske funksjonene forårsaket av leverdysfunksjon bidra til både initiering og progresjon av sepsis. Det retikuloendoteliale systemet i leveren fungerer som en første forsvarslinje for å rydde bakterier og deres produkter; leverdysfunksjon fører til et utslipp av disse produktene i systemisk sirkulasjon.
Leversvikt («sjokkert lever») kan manifestere seg ved forhøyede leverenzymer og bilirubin, koagulasjonsfeil og manglende utskillelse av giftstoffer som ammoniakk, som fører til forverret encefalopati.
Nyrefunksjon
Akutt nyreskade (AKI) ofte ledsaget sepsis. Forskjellige etiologier for AKI er rapportert, og årsaken antas vanligvis å være multifaktoriell. Mekanismen til AKI er kompleks, men involverer sannsynligvis en reduksjon i effektivt intravaskulært volum som skyldes systemisk hypotensjon, direkte renal vasokonstriksjon, frigjøring av cytokiner, og aktivering av nøytrofile stoffer med endotoksiner og andre peptider, som bidrar til nyreskade. De fleste dyreforsøk viser likevel at nyreblodstrømmen økes, ikke reduseres, i sepsis, men assosiert med nedsatt rørfunksjon og mangel på signifikant histologisk bevis på bular skade.
Sentralnervesystemets dysfunksjon
Involvering av sentralnervesystemet (CNS) i sepsis produserer encefalopati og perifer nevropati. Patogenesen er dårlig definert, men er sannsynligvis relatert til systemisk hypotensjon, noe som kan føre til hjernens hypoperfusjon.
Koagulopati
Subklinisk koagulopati, signalisert ved en mild forhøyning av trombintiden (TT) eller aktivert delvis tromboplastintid (aPTT) eller en moderat reduksjon i blodplateantallet, er ekstremt vanlig; Imidlertid kan åpen spredt intravaskulær koagulasjon (DIC) også utvikle seg. Protease-aktiverte reseptorer (PAR), spesielt PAR 1, danner den molekylære sammenhengen mellom koagulasjon og betennelse; PAR1 utøver cytobeskyttende effekter når det stimuleres av aktivert protein C eller lavdose-trombin, men utøver forstyrrende effekter på endotelcellebarrierefunksjon når det aktiveres av høydose trombin.
Mekanismer for organdysfunksjon og skade
De nøyaktige mekanismene for celleskade og resulterende organdysfunksjon i sepsis er ikke helt forstått. MODS er assosiert med utbredt endotel- og parenkymcelleskade, hvorav noen kan forklares med følgende 4 foreslåtte mekanismer.
Hypoksisk hypoksi
Septisk sirkulasjonslesjon forstyrrer oksygenering av vev, endrer metabolsk regulering av oksygentilførsel i vev og bidrar til organdysfunksjon. Mikrovaskulære og endotelavvik bidrar til septisk mikrosirkulasjonsdefekt i sepsis. De reaktive oksygenarter, lytiske enzymer og vasoaktive stoffer (f.eks. NO og endotelvekstfaktorer) fører til mikrosirkulasjonsskade, noe som er forsterket av erytrocyttenes manglende evne til å navigere i septisk mikrosirkulasjon. >
Direkte cytotoksisitet
Endotoksin, TNF-α og NO kan forårsake skade på mitokondriell elektrontransport, og føre til forstyrret energimetabolisme. Dette kalles cytopatisk eller histotoksisk anoksi, en manglende evne til å bruke oksygen selv når det er tilstede.
Apoptose
Apoptosis ( programmert celledød) er den viktigste mekanismen der dysfunksjonelle celler normalt blir eliminert. De proinflammatoriske cytokinene kan forsinke apoptose i aktiverte makrofager og nøytrofiler, men andre vev (f.eks. Tarmepitel) kan gjennomgå akselerert apoptose. Derfor spiller forstyrrelse av apoptose en kritisk rolle i vevskaden ved sepsis.
Immunosuppresjon
Interaksjonen mellom proinflammatorisk og betennelsesdempende meglere kan føre til ubalanse mellom dem. En inflammatorisk reaksjon eller en immunsvikt kan være dominerende, eller begge kan være tilstede.
Vertrespons og andre faktorer som påvirker utfallet
Kliniske egenskaper som er relatert til alvorlighetsgraden av sepsis inkluderer vertsrespons på infeksjon, infeksjonssted og type, tidspunkt og type antimikrobiell terapi, den fornærmende organismen, utvikling av sjokk, den underliggende sykdommen, pasientens langsiktige helsetilstand og antall mislykkede organer. Faktorer som fører til sepsis og septisk sjokk er kanskje ikke viktig for å bestemme det endelige resultatet.
Vertresponsen mot sepsis er preget av både proinflammatoriske responser og antiinflammatoriske immunsuppressive responser. Retningen, omfanget og varigheten av disse reaksjonene bestemmes av både vertsfaktorer (f.eks. Genetiske egenskaper, alder, sameksisterende sykdommer, medisiner) og patogenfaktorer (f.eks. Mikrobiell belastning, virulens).
Inflammatoriske responser initieres av interaksjon mellom patogenassosierte molekylære mønstre uttrykt av patogener og mønstergjenkjenningsreseptorer uttrykt av vertsceller på celleoverflaten (tolllignende reseptorer og C-type lektinreseptorer), i endosomet (TLRer), eller i cytoplasmaet (retinsyreinduserbart gen 1-lignende reseptorer og nukleotidbindende oligomeriseringsdomene-lignende reseptorer).
Konsekvensen av overdrevet betennelse er sikkerhetsvevskade og nekrotisk celledød, noe som resulterer i frigjøring av skadeassosierte molekylære mønstre, såkalte faremolekyler som vedvarer betennelse i det minste delvis ved å handle på de samme reseptorene for mønstergjenkjenning utløst av patogener.