Wprowadzenie
Od dziesięcioleci wiadomo, że interakcje między żywnością a lekami (BIZ) oraz zioła i leki ograniczają skuteczność leczenia. Ogromna liczba możliwych interakcji między wariacjami genetycznymi, trybami leczenia i licznymi związkami bioaktywnymi znajdującymi się w żywności i ziołach powoduje ogromną złożoność. Nowoczesne narzędzia, takie jak analiza dużych zbiorów danych, uczenie maszynowe i symulacja interakcji białko-ligand, mogą pomóc nam odpowiedzieć na cały zestaw pytań: Czy wybory żywieniowe mogą przyczynić się do niepowodzenia schematów terapeutycznych, a jeśli tak, to w jaki sposób? Jakie pokarmy należy spożyć przed przyjęciem przepisanego leku? I chyba najbardziej ekscytujące pytanie: jak możemy wykorzystać te narzędzia do przewidywania osobistych BIZ? Oczywiście, wiele odpowiedzi dotyczy metabolizmu leków, pokarmów i ziół przez cytochrom P450 3A4 (CYP3A4) w wątrobie i przewodzie pokarmowym (Galetin i in., 2010; Basheer i Kerem, 2015).
Większość genów kodujących enzymy CYP jest polimorficznych. Do chwili obecnej najbardziej wszechstronnym źródłem informacji dotyczących alleli CYP jest konsorcjum Pharmacogene Variation Consortium1, w którym reprezentowanych jest mniej niż 100 alleli CYP3A4. Spośród nich mniej niż 40 to egzoniczne SNP (polimorfizmy pojedynczego nukleotydu), których wynikiem jest zmodyfikowana sekwencja białka. Niewielka liczba osób we wszystkich wcześniej opublikowanych pracach na temat mutacji CYP3A4 dostarcza nam ograniczonych danych dotyczących rzeczywistych częstości występowania mutacji CYP3A4 w całej populacji i w określonych grupach.
Nie tylko, że wiarygodne informacje o występowaniu SNP są niekompletne , również ich implikacje kliniczne są w większości przypadków niejasne (Zanger i in., 2014). Zrozumienie, które i kiedy SNP mogą mieć znaczenie kliniczne, jest niezwykle złożonym zadaniem. Testy in vitro są czasochłonne, drogie i praktycznie nie mają większego znaczenia, biorąc pod uwagę dużą liczbę mutacji i nieskończoną liczbę kombinacji żywności i leków. Metody modelowania molekularnego, w tym obliczenia dokowania i wiązania wolnej energii, mogą służyć do przewidywania potencjalnego wpływu SNP i wielu związków na metabolizm, w którym pośredniczy CYP3A4 (Lewis i wsp., 1998). Na przykład oddziaływania niekowalencyjne, hydrofobowe, elektrostatyczne i van der Waalsa, wszystkie przyczyniają się do orientacji związku, a tym samym do jego wiązania i reakcji w aktywnym miejscu enzymu. To z kolei określi powinowactwo i specyficzność enzymu do różnych substratów oraz siłę inhibitorów enzymów (Kirchmair i in., 2012; Basheer i in., 2017).
Tutaj proponujemy nowy podejście do pomiaru częstości alleli mutacji CYP3A4 w różnych grupach etnicznych. To kompleksowe podejście ma moc uwidaczniania mutacji, które są powszechne w określonych grupach etnicznych, a w połączeniu z badaniami przesiewowymi pod kątem oddziałujących chemikaliów, np. Inhibitorów z pożywienia, pozwoli wyjaśnić wpływ poszczególnych mutacji na interakcję lek-żywność, służąc jako wstęp krok w kierunku medycyny spersonalizowanej i odżywiania. Ta praca może zwiększyć świadomość możliwego klinicznego znaczenia SNP CYP3A4 zmieniających białko, a także sugeruje kilka narzędzi niezbędnych do promocji i stosowania medycyny precyzyjnej i spersonalizowanej.
Materiały i metody
Przegląd bazy danych i analiza danych
Zbiór danych wariantów CYP3A4 został pobrany z przeglądarki gnomAD2 jako plik CVS. Do analizy i wizualizacji danych wykorzystano Python 2.7 z pakietami NumPy, pandas i matplotlib (patrz Dodatkowy arkusz danych S1). Aglomeracyjne hierarchiczne grupowanie przeprowadzono przy użyciu oprogramowania Expander 7 (Shamir i in., 2005) ze współczynnikiem korelacji rang Pearsona jako miarą podobieństwa i typu pełnego powiązania. Próg odległości ustalono na 0,6 dla grupowania SNP.
Modelowanie polimorfizmu in silico
Wydanie Maestro 2017-2 (Schrodinger, Nowy Jork, NY, Stany Zjednoczone) zostało wykorzystane do modelowanie obliczeniowe. Model dokowania CYP3A4 został zbudowany zgodnie z wcześniejszym opisem (Basheer i in., 2017). W skrócie, struktura krystaliczna CYP3A4 (pozycja PDB 2V0M) została przetworzona, zmodyfikowana i udoskonalona zgodnie z krokami Kreatora przygotowania białek. Siatka dokująca z metalowym ograniczeniem koordynacji dla Fe2 + w grupie hemu została wygenerowana na podstawie centroidu ketokonazolu w pierwotnym miejscu wiązania w strukturze kryształu. Do symulacji dokowania wybrano siedem mutacji, po jednej reprezentatywnej dla każdej grupy etnicznej (tabele 1, 2). Dla każdego wariantu białka przed etapami przygotowania białka wprowadzono pojedynczą mutację punktową. Struktury 3D ligandów zostały wygenerowane na podstawie struktur 2D z PubChem3 i przygotowane do dokowania za pomocą zadania LigPrep.Pole siłowe OPLS3 i domyślne opcje Szybowania dla standardowej precyzji zostały zastosowane w modelu dokowania, z wyjątkiem tego, że zastosowano ograniczenie koordynacji metalu, a także 30 pozycji dla liczby póz do uwzględnienia i 10 pozycji dla liczby pozycji do zapisania na zewnątrz. Dla każdego liganda wybrano wynik dokowania z najniższym wynikiem emodelu Glide.

Tabela 1. Wybrane reprezentatywne SNP dla siedmiu grup etnicznych.

Tabela 2. Częstość (%) wybranych mutacji według grupy etnicznej.
Wyniki
Baza danych agregacji genomu (gnomAD; patrz przypis 2) gromadzi dane dotyczące sekwencjonowania zarówno egzomu, jak i genomu z wielu różnych projektów sekwencjonowania na dużą skalę. Obejmuje dane z 125 748 sekwencji egzomu i 15 708 sekwencji całego genomu od 141 456 niespokrewnionych osobników reprezentujących siedem populacji etnicznych (Lek i in., 2016). Baza danych GnomAD prezentuje 856 wariantów CYP3A4, z których 397 to intronowe, a aż 459 to egzoniczne. Spośród egzonowych SNP 312 to mutacje typu missense, co wskazuje, że wpływają one na strukturę białka. Gen CYP3A4 ma długość 34 205 pz. Jego 13 eksonów zawiera region kodujący o długości 1512 pz, który wytwarza białko o 504 aminokwasach. 412 egzonicznych SNP z unikalnymi pozycjami w tym genie skutkuje gęstością egzonową SNP wynoszącą 272 / kbp (tabela uzupełniająca S1).
Obliczenie zróżnicowanej częstości alleli na grupę etniczną ujawnia, że niektóre populacje wykazują wyższą częstość mutacji (Rysunek 1A). Większość mutacji CYP3A4 w populacji europejskiej jest rzeczywiście rzadka, jak się powszechnie uważa, podczas gdy mutacje w innych populacjach, takich jak afrykańska i wschodnioazjatycka, są znacznie bardziej rozpowszechnione (tabela uzupełniająca S2).
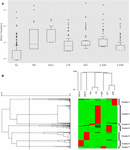
Rysunek 1. Analiza SNP typu missense CYP3A4 w siedmiu różnych populacjach. (A) Wykres skrzynkowy w skali logarytmicznej częstości alleli. Ramki przedstawiają przedział międzykwartylowy (IQR), niebieskie linie przedstawiają mediany, wąsy przedstawiają dane w granicach 1,5 IQR, a wartości odstające są pokazane jako małe kółka. (B) Hierarchiczne grupowanie częstotliwości allelicznych. Każdy wiersz reprezentuje pojedynczy SNP. Każda kolumna przedstawia odrębną populację etniczną. Częstość alleli SNP w każdej z populacji jest reprezentowana przez kolor odpowiedniej komórki w pliku macierzy. Zielony i czerwony reprezentują odpowiednio niską i wysoką częstotliwość. Górny dendrogram pokazuje podobieństwa we wzorcu częstotliwości alleli między każdą grupą badanych. Lewy dendrogram przedstawia skupienie genów w dwóch grupach. Linia przerywana przedstawia próg odległości 0,6 używany do podziału na grupy. UE – europejski (niefiński; n = 64603), FIN – europejski (fiński; n = 12562), ASH J – aszkenazyjski (n = 5185), LTN – latynoski (n = 17720), AFR – afrykański (n = 12 487), E ASN – Azja Wschodnia (n = 9 977), S ASN – Azja Południowa (n = 64 603).
Użyliśmy hierarchicznego grupowania, aby pogrupować warianty o podobnych wzorcach częstotliwości. Nasza analiza danych dostarczyła siedmiu różnych klastrów (Rysunek 1B). Ponadto wyraźnie widać, że SNP o wysokiej częstotliwości w każdym klastrze są charakterystyczne dla jednej określonej populacji. Hierarchiczna analiza skupień grup etnicznych wspiera powiązanie między zmiennością genetyczną a pochodzeniem etnicznym poprzez grupowanie razem pokrewnych grup etnicznych, takich jak mieszkańcy Azji Południowej i Wschodniej, a także Europejczycy z Finlandii i spoza Finlandii.
Do oceny zastosowano model obliczeniowy możliwy wpływ mutacji punktowych w CYP3A4 na jego zdolność do wiązania substratów i inhibitorów. CYP3A4 jest zdolny do utleniania wielu związków endogennych i ksenobiotyków. Tutaj ketokonazol został wybrany jako reprezentatywny lek i bardzo skuteczny specyficzny inhibitor; androstendion i testosteron wybrano jako reprezentatywne endogenne hormony; a demetoksykurkuminę i epigallokatechinę wybrano jako przedstawicieli dietetycznych substancji bioaktywnych. Zbudowano model dokowania do przewidywania pozycji wiązania wybranych związków w miejscu wiązania CYP3A4. Model został po raz pierwszy zweryfikowany poprzez pomyślne przywrócenie pozycji ketokonazolu w miejscu wiązania z RMSD 1,52 Å w stosunku do oryginalnej struktury kryształu. Siedem zmutowanych białek zaprojektowano w oparciu o strukturę krystaliczną białka typu dzikiego (rysunek uzupełniający S1). Dla każdej grupy etnicznej jako reprezentatywna wybrano najczęstszą unikalną mutację. Wpływ pojedynczych mutacji na wiązanie substratu oceniano na podstawie porównania między pozycjami dokowania na natywnym białku i na wariantach białek. Zmiany w pozycjach dokowania pod względem RMSD podsumowano w Tabeli 3.

Tabela 3. RMSD względem WT łączących ligandów z miejscami wiązania siedmiu wariantów CYP3A4.
Stwierdzono, że wpływ SNP CYP3A4 na wiązanie substratu jest specyficzny dla mutacji. Tylko w nielicznych przypadkach mutacje spowodowały zmianę pozycji wiązania liganda w kieszeni wiążącej. Pozycja dokująca testosteronu była taka sama we wszystkich siedmiu testowanych wariantach. Warianty E262K, D174H i K168N nie spowodowały zmiany pozycji wiązania w żadnej z badanych cząsteczek. Jednak mutacje L373F i T163A zmieniły pozycję wiązania androstendionu, tak że był on umieszczony równolegle do grupy hemu, a nie prostopadle do niej, jak w białku WT. Również androstendion poddano rotacji tak, że grupa cyklopentanonowa znajduje się proksymalnie do hemu, zamiast grupy cykloheksanonu w białku WT. Mutacje S222P i L293P powodowały jedynie niewielką rotację w pozycji wiązania androstendionu (ryc. 2A). Spośród wszystkich badanych mutacji tylko S222P powodował istotne zmiany w pozycjach dokowania ketokonazolu i demetoksykurkuminy w miejscu wiązania (ryc. 2B, C); podczas gdy w przypadku epigallokatechiny mutacją powodującą zmianę pozy była L373F (Rysunek 2D).
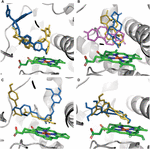
Rysunek 2. Modele ligandów zadokowanych w miejscu wiązania CYP3A4. (A) Ketokonazol, (B) androstenodion, (C) demetoksykurkumina i (D) epigallokatechina. Miejsce wiązania białka jest reprezentowane przez szare wstążki; Hem jest reprezentowany przez zielone patyki, pozycje dokowania w białku WT, a mutanty S222P i L373F są pokazane odpowiednio jako pomarańczowe, niebieskie i fioletowe patyki. Pozycje dokowania Androstendionu w wariantach L293P i T136A nakładają się odpowiednio na pozy w wariantach S222P i L373F.
Dyskusja
Cytochrom P450 3A4 jest głównym enzymem odpowiedzialnym za interakcje między żywnością a lekami. Aktualne badania mutacji w CYP3A4 koncentrowały się na kilkudziesięciu SNP znalezionych w wyznaczonych badaniach (Sata i wsp., 2000; Dai i wsp., 2001; Eiselt i wsp., 2001; Hsieh i wsp., 2001; Lamba i wsp. ., 2002; Murayama i wsp., 2002). Jak wykazano tutaj, reprezentują one wierzchołek góry lodowej, biorąc pod uwagę częstość występowania i potencjalne skutki mutacji CYP3A4. Mnogość projektów sekwencjonowania dużych genomów i egzomów otworzyła nową drogę do identyfikacji wielu nieznanych mutacji. Tutaj pokazujemy, że wcześniej przedstawione mutacje są tylko wierzchołkiem góry lodowej, demonstrując 856 mutacji występujących w CYP3A4, z których jedna trzecia modyfikuje strukturę białka. Na podstawie kohorty 141 456 niespokrewnionych osób obliczono dokładne częstości alleliczne mutacji CYP3A4 dla siedmiu różnych grup etnicznych. Według naszej najlepszej wiedzy, jest to największe i najbardziej wszechstronne badanie z dużą ilością danych, dotyczące mutacji egzonicznych CYP3A4 i częstości ich alleli w różnych populacjach, opublikowane do tej pory.
Polimorficzne enzymy CYP3A4 mogą być bardzo ważne w wyjaśnianiu różnice w skuteczności i toksyczności leków u różnych osób. Mutacje w genie CYP3A4 mogą prowadzić do zniesienia, zmniejszenia, zmiany lub zwiększenia aktywności enzymatycznej. Mutacje egzoniczne mogą modyfikować aktywność enzymatyczną, co wykazano w kilku badaniach klinicznych z wybranymi substratami. Niektóre przypadki zmienionego metabolizmu z powodu SNP w CYP3A4 zostały już opisane w literaturze (Eiselt i in., 2001; Miyazaki i in., 2008). Pomimo funkcjonalnego znaczenia i klinicznego znaczenia SNP w CYP3A4 i prawdopodobnie ze względu na ich stosunkowo niską zidentyfikowaną częstość w populacji ogólnej, polimorfizm CYP3A4 nie wzbudził takiej uwagi, na jaką zasługuje.
W tym przypadku siedem mutacji posłużyło do przewidywania wpływ SNP na orientację wiązania substratu i inhibitora. W literaturze polimorfizm CYP3A4 dzieli populację ogólną na trzy grupy – słabo metabolizujący, normalnie metabolizujący i szybko metabolizujący, w oparciu o intronowe SNP, które modyfikują poziomy ekspresji, a nie strukturę (Zanger i Schwab, 2013). Nasze obliczenia sugerują dodatkową klasyfikację: zmieniony metabolizm. Niektóre mutacje zaproponowane przez nasz wirtualny model spowodowałyby zmianę orientacji wiązania poszczególnych ligandów. Oczekuje się, że zmiany te zmniejszą prawdopodobieństwo enzymatycznego utleniania z powodu zwiększonej odległości od hemu lub doprowadzą do powstania produktów, które w przeciwnym razie nie byłyby widoczne podczas testów toksyczności przeprowadzanych w ramach procesu opracowywania leku. Jednak, jak przewiduje nasz model, dla większości substratów mutacje CYP3A4 są łagodne.
Zmodyfikowana pozycja substratu w kieszeni wiążącej z powodu zmiany strukturalnej białka to tylko jeden możliwy mechanizm, dzięki któremu mutacja może zmienić aktywność białka . Upośledzone zakotwiczenie białka w błonie, uszkodzone kanały prowadzące przez substrat i upośledzone wyjście produktów stanowią dodatkowe mechanizmy mutacyjnej zmiany aktywności białka. Jak pokazano tutaj, efekt każdej mutacji jest specyficzny dla substratu.Ustalenie, które kombinacje substratów i mutacji mogą modyfikować aktywność enzymatyczną przy użyciu tradycyjnych metod in vitro jest pracochłonne, podkreślając potrzebę predykcyjnych narzędzi wirtualnych w rozwiązywaniu tej złożonej zagadki.
Publiczne i zawodowe zainteresowanie medycyną osobistą i precyzyjną szybko rośnie. Przewidywanie zmodyfikowanego metabolizmu leków na podstawie indywidualnego polimorfizmu CYP3A4 wydaje się być tylko kwestią czasu. Tutaj proponujemy, aby różne grupy etniczne miały unikalne zestawy SNP CYP3A4. W istocie pochodzenie etniczne może służyć jako pierwszy wykonalny krok w medycynie spersonalizowanej, poprzedzający wdrożenie indywidualnego badania DNA dla wszystkich. Co ciekawe, pochodzenie etniczne ma jeszcze jeden wpływ na metabolizm leków przez CYP3A4, będąc głównym czynnikiem w określaniu wyborów żywieniowych i nawyków żywieniowych. Można zasugerować, że schematy terapeutyczne powinny być specjalnie zaprojektowane dla każdej grupy etnicznej, przynajmniej dla leków, które są silnie metabolizowane przez CYP3A4. Podkreśla to możliwości wykorzystania i integracji baz danych oraz głębokiego uczenia się w celu określenia, w jaki sposób SNP, pochodzenie etniczne, składniki diety i leki modyfikują aktywność CYP3A4 i powodzenie leczenia.
Dostępność danych
W tym badaniu przeanalizowano publicznie dostępne zbiory danych. Te dane można znaleźć tutaj: http://gnomad.broadinstitute.org/gene/ENSG00000160868.
Wkład autorów
Wszyscy wymienieni autorzy wnieśli znaczący, bezpośredni i intelektualny wkład w pracy i zatwierdzili je do publikacji.
Oświadczenie o konflikcie interesów
Autorzy oświadczają, że badanie zostało przeprowadzone przy braku jakichkolwiek powiązań handlowych lub finansowych, które można by zinterpretować jako potencjalne konflikt interesów.
Materiały dodatkowe
RYSUNEK S1 | Model wstęgowy 3D CYP3A4 i lokalizacja zmutowanych aminokwasów w siedmiu wariantach białek zaprojektowanych do dokowania. Hem jest reprezentowany jako zielone patyki, Fe2 + jako czerwona kula, SNP używane w analizie in silico są reprezentowane jako czerwone obszary na wstążce, a grupy R zmutowanych aminokwasów w modelach wariantowych są wyraźnie widoczne jako jasnoszare patyki.
TABELA S1 | Typy SNP CYP3A4 w populacji 141 456 niespokrewnionych osobników reprezentujących 7 populacji etnicznych.
TABELA S2 | SNP CYP3A4 według grup etnicznych.
Przypisy
- ^ www.pharmvar.org
- ^ https://gnomad.broadinstitute.org
- ^ https://pubchem.ncbi.nlm.nih.gov