Zespół Angelmana (AS) to rzadkie zaburzenie neurogenetyczne, które dotyka mniej więcej 1 na 15 000 osób – około 500 000 osób na całym świecie. Dzieci i dorośli z ZA zazwyczaj mają problemy z równowagą, upośledzeniem motorycznym i osłabiającymi drgawkami. Niektóre osoby nigdy nie chodzą. Większość nie mówi. Zakłócone cykle snu mogą również być poważnym wyzwaniem dla jednostki i opiekunów. Osoby z ZA wymagają ciągłej opieki i nie są w stanie żyć samodzielnie. Mają normalną długość życia. Takie jest dzisiejsze życie ludzi z zespołem Angelmana, ale nadzieja jest tutaj. Naukowcy uważają, że AS ma największy potencjał do wyleczenia w porównaniu z innymi zaburzeniami neurogenetycznymi, a FAST (Foundation for Angelman Syndrome Therapeutics) ma bardzo zaawansowany plan, aby to osiągnąć.
Typy Przyczyny Testy & Materiały diagnostyczne
Rodzaje zespołu Angelmana
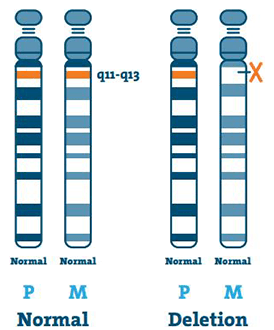
Usunięcie ( 65-75%)
DNA (kwas dezoksyrybonukleinowy) jest głównym składnikiem chromosomów. Zawiera nasz unikalny kod genetyczny. U większości osób z ZA brakuje fragmentu DNA w regionie 15q11-13 na matczynym chromosomie 15.
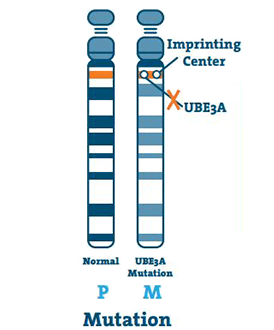
Mutacja (5 -11%)
Dzieje się tak, gdy występuje niewielka nieprawidłowość w DNA genu UBE3A. Mutacja może wystąpić w dowolnym miejscu genu.
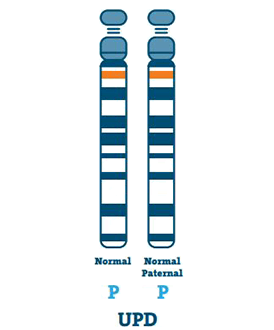
Disomia rodzicielska (3-7%)
Osoba z UPD ma dwie kopie chromosomu 15 od ojca, zamiast po jednej od ojca i matki. UPD zwykle występuje, gdy w jajku nie ma chromosomu 15.
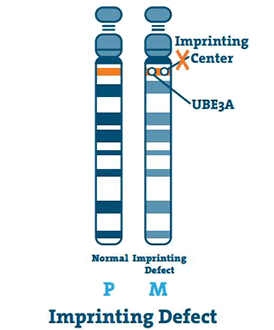
Defekt nadruku (< 3%)
Centrum nadruku to mały odcinek DNA znajdujący się w regionie q11-13 chromosomu. W rzadkich przypadkach chromosom 15 matki jest pusty, a środek kopiuje chromosom 15 ojca. Jest to wada odciskowa.
Charakterystyka
Typowe cechy ZZSK zwykle nie są widoczne narodziny. Osoby z tym zaburzeniem mają trudności z karmieniem jako niemowlęta i zauważalne opóźniony rozwój w wieku około 6-12 miesięcy. Potrzebują intensywnych terapii, aby rozwinąć umiejętności funkcjonalne. AS wpływa na każdą rasę i obie płcie. Często jest błędnie diagnozowany jako autyzm lub porażenie mózgowe.
Cechy behawioralne
Osoby z zespołem Angelmana mają pewne wyraźne cechy behawioralne, w tym radosne zachowanie, charakteryzujące się częstym śmiechem, uśmiechem i pobudliwością. Wiele osób z ZA fascynuje się wodą i czerpie wielką przyjemność z takich czynności, jak pływanie i kąpiel.
Przyczyny zespołu Angelmana
Zespół Angelmana to zaburzenie jednogenowe spowodowane utratą funkcji w genie UBE3A na 15 chromosomie matki. Ludzie mają dwa zestawy chromosomów – jeden odziedziczony po matce i jeden od ojca. U typowej osoby gen UBE3A odziedziczony po matce jest aktywny, podczas gdy kopia genu odziedziczonego po ojcu jest wyciszana w neuronach w naszych mózgach – zjawisko to nosi nazwę imprintingu. W przypadku osób z ZA ten matczyny gen nie spełnia swojego zadania, a to wpływa na ich Messenger RNA (mRNA).
![]()
Co to jest mRNA?
mRNA to FedEx ciała. Nasze DNA wykorzystuje mRNA jako usługę dostawczą do wysyłania planów do fabryk montażu białek w naszych komórkach. Osoby z ZA mają mutację, delecję lub inny defekt w swoim genie UBE3A, który przerywa tę usługę dostarczania. W rezultacie ich neurony nie wytwarzają żadnego funkcjonalnego białka UBE3A i to właśnie wyzwala objawy ZA. To białko pomaga nam chodzić, rozmawiać i wykonywać inne codzienne czynności.
![]()
Czy to genetyka?
W większości przypadków zespół Angelmana nie jest dziedziczony – szczególnie te spowodowane delecją lub UPD. Zamiast tego te zmiany genetyczne występują jako zdarzenia losowe podczas tworzenia się komórek rozrodczych lub we wczesnym rozwoju embrionalnym.
Testy & Diagnoza
Dowiedz się o kryteriach, według których lekarze określają, czy dana osoba ma zespół Angelmana, i poczytaj o dostępnych testach i częstych błędnych diagnozach.
![]()
Testowanie
![]()
Diagnoza

Dołącz do naszej społeczności
Nawiąż kontakt z innymi osobami, które mają dziecko lub ukochaną osobę z zespołem Angelmana.
