Angelmans syndrom (AS) är en sällsynt neurogenetisk störning som drabbar ungefär en av 15 000 personer – cirka 500 000 individer världen över. Barn och vuxna med AS har vanligtvis balansproblem, motorisk försämring och försvagande anfall. Vissa individer går aldrig. De flesta talar inte. Störda sömncykler kan också vara en allvarlig utmaning för individen och vaktmästaren. Individer med AS behöver kontinuerlig vård och kan inte leva självständigt. De har en normal livslängd. Detta är livet idag för människor som lever med Angelmans syndrom, men hoppet är här. Forskare tror att AS har den största potentialen för att bli botad jämfört med andra neurogenetiska störningar, och FAST (Foundation for Angelman Syndrome Therapeutics) har en plan på god väg för att uppnå just det.
Typer Orsaker Tester & Diagnosresurser
Typer av Angelmans syndrom
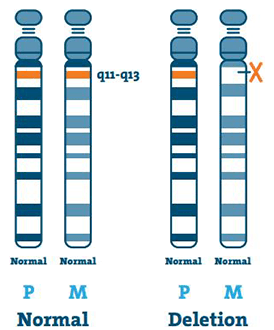
Radering ( 65-75%)
DNA (deoxiribonukleinsyra) är huvudkomponenten i kromosomer. Den innehåller vår unika genetiska kod. De flesta individer med AS saknar en bit DNA i region 15q11-13 på moderns kromosom 15.
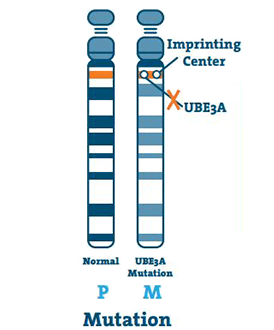
Mutation (5 -11%)
Detta inträffar när det finns en liten abnormitet i DNA från UBE3A-genen. En mutation kan inträffa var som helst på genen.
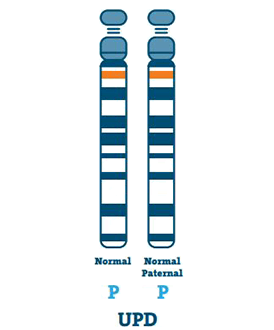
Uniparental Disomy (3-7%)
En person med UPD har två kopior av kromosom 15 från sin far, i stället för en vardera från fadern och mamman. UPD händer vanligtvis om det inte finns någon kromosom 15 i ägget.
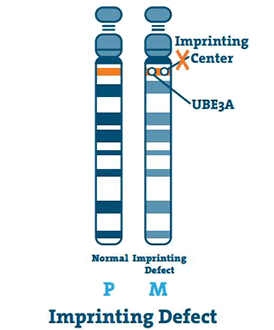
Imprinting Defect (< 3%)
Avtryckscentret är en liten DNA-sträcka som ligger i kromosomens Q11-13-region. I sällsynta fall är moderns kromosom 15 tom och mitten kopierar faderns kromosom 15. Detta är en präglingsfel.
Egenskaper
Typiska egenskaper hos AS är vanligtvis inte uppenbara vid födelse. Personer med sjukdomen har matningssvårigheter som spädbarn och märkbar fördröjd utveckling runt 6-12 månaders ålder. De behöver intensiva terapier för att utveckla funktionella färdigheter. AS påverkar varje lopp och båda könen. Det diagnostiseras ofta felaktigt som autism eller cerebral pares.
Beteendeegenskaper
Människor med Angelmans syndrom har vissa distinkta beteendeegenskaper, inklusive en glad uppträdande, som kännetecknas av ofta skratt, leende och upphetsning. Många individer med AS har en fascination av vatten och tycker mycket om aktiviteter som bad och bad.
Orsaker till Angelmans syndrom
Angelmans syndrom är en en-genstörning orsakad av förlust. funktion i UBE3A-genen på moderns 15: e kromosom. Människor har två uppsättningar kromosomer – en ärvd från modern och en från fadern. I en typisk person är den modernt ärvda UBE3A-genen aktiv, medan kopian av genen som ärvs från fadern tystas i nervcellerna i våra hjärnor – ett fenomen som kallas imprinting. För personer med AS gör denna modergen inte sitt jobb, och det påverkar deras Messenger-RNA (mRNA).
![]()
Vad är mRNA?
mRNA är kroppens FedEx. Vårt DNA använder mRNA som en leveransservice för att skicka ritningar till proteinmonteringsfabrikerna i våra celler. Människor med AS har en mutation, radering eller annan defekt i sin UBE3A-gen som avbryter denna leveransservice. Som ett resultat gör deras neuroner inte något funktionellt UBE3A-protein, och det är det som utlöser symtomen på AS. Detta protein är det som hjälper oss att gå, prata och utföra andra vardagliga uppgifter.
![]()
Är det genetiskt?
I de flesta fall ärvs inte Angelmans syndrom – särskilt de som orsakas av en radering eller UPD. Istället inträffar dessa genetiska förändringar som slumpmässiga händelser under bildandet av reproduktiva celler eller i tidig embryonal utveckling.
Tester & Diagnos
Lär dig om kriterierna som läkare använder för att avgöra om en individ har Angelmans syndrom och läsa om tillgängliga tester och vanliga feldiagnoser.
![]()
Testning
![]()
Diagnos

Gå med i vår community
Anslut till andra personer som har ett barn eller älskade med Angelmans syndrom.
