Malign intravaskulär inflammation
Sepsis har kallats en process för malign intravaskulär inflammation . Normalt säkerställer en potent, komplex, immunologisk kaskad ett snabbt skyddande svar på mikroorganisminvasion hos människor. Ett bristfälligt immunologiskt försvar kan tillåta infektion att etablera sig; emellertid kan ett överdrivet eller dåligt reglerat svar skada värden genom otillräcklig frisättning av inhemskt genererade inflammatoriska föreningar (se bilden nedan).
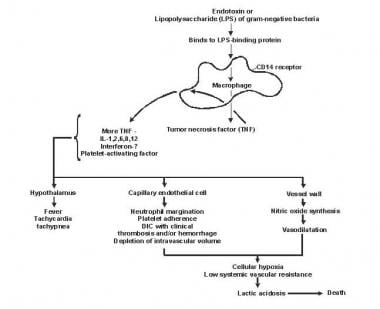 Patogenes av sepsis och multiorgansvikt.
Patogenes av sepsis och multiorgansvikt. Lipid A och andra bakteriella produkter frisätter cytokiner och andra immunmodulatorer som förmedlar de kliniska manifestationerna av sepsis. Interleukiner, tumörnekrosfaktor (TNF) -α, interferon gamma (IFN-γ) och andra kolonistimulerande faktorer produceras snabbt inom några minuter eller timmar efter interaktioner mellan monocyter och makrofager med lipid A.
Inflammatorisk mediatorfrisättning blir en självstimulerande process och frisättning av andra sådana mediatorer, inklusive interleukin (IL) -1, trombocytaktiverande faktor, IL-2, IL-6, IL-8, IL-10 och kväveoxid (NO) ökar ytterligare cytokinnivåerna. Detta leder till fortsatt aktivering av polymorfonukleära leukocyter (PMN), makrofager och lymfocyter; proinflammatoriska medlare rekryterar fler av dessa celler. Alla dessa processer skapar ett tillstånd av destruktiv immunologisk dissonans.
Sepsis beskrivs som en autodestruktiv process som möjliggör förlängning av det normala patofysiologiska svaret på infektion för att involvera annars normala vävnader och resultat i MODS. Organdysfunktion eller organsvikt kan vara det första kliniska tecknet på sepsis, och inget organsystem är immunt från konsekvenserna av inflammatoriska överdriven sepsis. Dödligheten ökar när organsvikt ökar.
Även om okontrollerat, när MODS utvecklar systemiska bevis för både proinflammatorisk och antiinflammatorisk uppreglering, är vanligtvis närvarande, vilket tyder på att misslyckande av värdförsvarshomeostas är den sista vägen från sepsis till MODS, snarare än enkel hypotensioninducerad slutorganskada, vilket kan uppstå vid hemorragisk chock. Överlevnad från svår sepsis med MODS är vanligtvis associerad med en generaliserad minskning av både det proinflammatoriska och antiinflammatoriska svaret.
En ny hypotes har nyligen framkommit att överlevnad från svår sepsis kräver en generaliserad nedreglering av kroppens immunsvar, energiska funktioner och tillhörande organprestanda. Således kan MODS genom värdens adaptiva svar på överväldigande inflammation, vilket gör att inflammation kan rensas utan att orsaka permanent slutorganskada. Som diskuteras nedan avslöjar alla organ en generaliserad hyporesponsivitet som är klart onormal i hälsan men kan markera en överlevnadsstrategi vid svår sepsis.
Dysfunktion i organsystem
Cirkulationsstörningar
Betydande störningar vid automatisk reglering av cirkulationen är typiskt för sepsis. Vasoaktiva medlare orsakar vasodilatation och ökar mikrovaskulär permeabilitet på infektionsstället. NO spelar en central roll i vasodilatationen av septisk chock. Dessutom kan nedsatt utsöndring av vasopressin förekomma, vilket kan möjliggöra bestående vasodilatation.
Förändringar i både systolisk och diastolisk ventrikulär prestanda uppträder i sepsis. Genom att använda Frank-Starling-mekanismen ökar hjärtproduktionen ofta för att upprätthålla blodtrycket i närvaro av systemisk vasodilatation. Patienter med redan existerande hjärtsjukdomar kan inte öka sin hjärtproduktion på lämpligt sätt.
Regionalt stör sepsis den normala fördelningen av systemiskt blodflöde till organsystem. Följaktligen kanske kärnorgan inte får lämplig syretillförsel, och resultatet är det som kallas regional hypoperfusion.
Mikrocirkulation är det viktigaste målorganet för skada vid sepsis eftersom vaskulärt endotel är påverkas universellt av de cirkulerande inflammatoriska medlarna. Även om det är oklart om mikrocirkulationsavvikelser är orsaken eller en oskyldig åskådare av organskadorna, sågs tydlig mikrovaskulär dysfunktion. En minskning av antalet perfunderade kapillärer ses, men vid tillämpning av vasodilaterande behandlingar sker full mikrovaskulär rekrytering. Mitokondriell dysfunktion uppträder också och är ofta associerad med minskade mitokondriella transmembranpotentialgradienter, vilka är nödvändiga för att driva oxidativ fosforylering. Slutresultatet är en uppenbar oförmåga hos slutorganen att extrahera syre maximalt.
Debatten fortsätter om detta misslyckande med energimetabolism är en adaptiv cytoprotektiv mekanism som liknar viloläge eller återspeglar primär mitokondriell patologi. Dessa är områden för aktiv forskning men översätts för närvarande inte till tydliga riktlinjer för klinisk praxis. Ökad kapillär endotelpermeabilitet leder till utbredd proteinrik vävnadsödem.
Septisk chock och SIRS kännetecknas av reversibel hjärtinfarktdepression, vilket kan visa sig resistent mot katekolamin och vätskeadministrering. Cirkulerande ”myocardial depressant factor” – som antagligen representerar de synergistiska effekterna av TNF-α, IL-1β, andra cytokiner och NO – är inblandad i patogenes. De två egenskaperna hos denna akuta stressmyokardiala depression är försämrad adrenerg respons och diastolisk dysfunktion som leder till relativ katekolaminresistens och små snarare än utvidgade hjärtan. Makrovaskulär myokardisk iskemi och hypoperfusion är osannolika bidragande.
Vid svår sepsis och septisk chock orsakar mikrocirkulationsdysfunktion och mitokondriell depression regional vävnadsnöd, och regional dysoxi kvarstår därför. Detta tillstånd kallas mikrocirkulations- och mitokondriellt nödsyndrom (MMDS). Sepsisinducerad inflammatorisk autoregulatorisk dysfunktion kvarstår och syrebehovet matchas inte av tillförsel, vilket leder till MODS.
Omfördelning av intravaskulär vätskevolym till följd av minskad arteriell vaskulär ton, minskad venös återkomst från venös di lation och frisättning av hjärtinfarktämnen orsakar hypotoni.
Pulmonell dysfunktion
Endotelskada i lungkärl leder till störningar kapillärblodflöde och förbättrad mikrovaskulär permeabilitet, vilket resulterar i interstitiellt och alveolärt ödem. Neutrofil infångning i lungmikrocirkulationen initierar och förstärker skadan på alveolära kapillärmembran. Akut lungskada och akut andningsnedsyndrom (ARDS) är frekventa manifestationer av dessa effekter. Sepsis och lunginflammation är faktiskt de vanligaste orsakerna till ARDS.
Gastrointestinal dysfunktion
Mage-tarmkanalen (GI) kan hjälpa till att sprida sepsisskada. Överväxt av bakterier i övre mag-tarmkanalen kan sugas in i lungorna, vilket ger nosokomiell eller aspirationspneumoni. Tarmens normala barriärfunktion kan påverkas, vilket möjliggör translokation av bakterier, endotoxiner och normala matsmältningsproteaser i den systemiska cirkulationen och förlänger septiskt svar.
Septisk chock kan orsaka paralytisk ileus som kan leda till en fördröjning av instruktionen för enteral utfodring. Överskott av NO-produktion anses vara det orsakande medlet för sepsisinducerad ileus. Den optimala nivån av näringsintag störs inför höga protein- och kaloribehov. Narkotika och muskelavslappnande medel kan ytterligare förvärra rörligheten i mag-tarmkanalen.
Leverdysfunktion
Som en följd av den roll levern spelar i värdförsvar kan de onormala syntetiska funktionerna orsakade av leverdysfunktion bidra till både initiering och progression av sepsis. Levers retikulo-endotel-system fungerar som en första försvarslinje för att rensa bakterier och deras produkter; leversvikt leder till ett utsläpp av dessa produkter till systemisk cirkulation.
Leversvikt (”chockad lever”) kan manifesteras av förhöjningar i leverenzymer och bilirubin, koagulationsdefekter och underlåtenhet att utsöndra toxiner som ammoniak, vilket leder till försämrad encefalopati.
Njurfunktion
Akut njurskada (AKI) ofta åtföljer sepsis. Olika etiologier för AKI har rapporterats och orsaken anses typiskt vara multifaktoriell. Mekanismen för AKI är komplex men involverar sannolikt en minskning av effektiv intravaskulär volym till följd av systemisk hypotoni, direkt njur vasokonstriktion, frisättning av cytokiner och aktivering av neutrofiler av endotoxiner och andra peptider, vilket bidrar till njurskada. Ändå visar de flesta djurstudier att njurblodflödet är ökat, inte minskat, i sepsis, men associerat med nedsatt rörfunktion och brist på signifikant histologiskt bevis på tu bular skada.
Dysfunktion i centrala nervsystemet
Involvering av centrala nervsystemet (CNS) i sepsis ger encefalopati och perifer neuropati. Patogenesen är dåligt definierad men är troligtvis relaterad till systemisk hypotoni, vilket kan leda till hjärnhypoperfusion.
Koagulopati
Subklinisk koagulopati, signalerad genom en mild höjning av trombintiden (TT) eller aktiverad partiell tromboplastintid (aPTT) eller en måttlig minskning av trombocytantalet är extremt vanligt; emellertid kan öppen disseminerad intravaskulär koagulation (DIC) också utvecklas. Proteasaktiverade receptorer (PAR), särskilt PAR 1, bildar den molekylära länken mellan koagulation och inflammation; PAR1 utövar cytoprotektiva effekter när det stimuleras av aktiverat protein C eller lågdos-trombin men utövar störande effekter på endotelcellbarriärfunktionen när det aktiveras av högdos-trombin.
Mekanismer för organdysfunktion och skada
De exakta mekanismerna för cellskada och resulterande organdysfunktion vid sepsis är inte helt förstådda. MODS är associerad med utbredd endotel- och parenkymcellskada, varav några kan förklaras med följande fyra föreslagna mekanismer.
Hypoxisk hypoxi
Septisk cirkulationsskada stör vävnads syresättning, förändrar metabolisk reglering av vävnads syretillförsel och bidrar till organdysfunktion. Mikrovaskulära och endotelavvikelser bidrar till septisk mikrocirkulationsfel i sepsis. De reaktiva syrearten, lytiska enzymer och vasoaktiva substanser (t.ex. NO- och endoteltillväxtfaktorer) leder till mikrocirkulationsskada, vilket förvärras av erytrocyternas oförmåga att navigera i septisk mikrocirkulation.
Direkt cytotoxicitet
Endotoxin, TNF-α och NO kan orsaka skador på mitokondriell elektrontransport, vilket leder till störd energimetabolism. Detta kallas cytopatisk eller histotoxisk anoxi, en oförmåga att använda syre även när det är närvarande.
Apoptos
Apoptos ( programmerad celldöd) är den huvudsakliga mekanismen genom vilken dysfunktionella celler normalt elimineras. De proinflammatoriska cytokinerna kan fördröja apoptos i aktiverade makrofager och neutrofiler, men andra vävnader (t ex tarmepitel) kan genomgå accelererad apoptos. Därför spelar störning av apoptos en avgörande roll i vävnadsskada vid sepsis.
Immunsuppression
Interaktionen mellan proinflammatorisk och antiinflammatoriska medlare kan leda till en obalans mellan dem. En inflammatorisk reaktion eller en immunbrist kan dominera, eller båda kan vara närvarande.
Värdrespons och andra faktorer som påverkar resultatet
Kliniska egenskaper som relaterar till svårighetsgraden av sepsis inkluderar värdrespons på infektion, plats och typ av infektion, tidpunkten och typen av antimikrobiell terapi, den kränkande organismen, utvecklingen av chock, den underliggande sjukdomen, patientens långvariga hälsotillstånd och antalet misslyckade organ. Faktorer som leder till sepsis och septisk chock kanske inte är nödvändiga för att bestämma det slutliga resultatet.
Värdsvaret mot sepsis kännetecknas av både proinflammatoriska svar och antiinflammatoriska immunsuppressiva svar. Riktning, omfattning och varaktighet för dessa reaktioner bestäms av både värdfaktorer (t.ex. genetiska egenskaper, ålder, samexisterande sjukdomar, mediciner) och patogenfaktorer (t.ex. mikrobiell belastning, virulens).
Inflammatoriska svar initieras av interaktion mellan patogenassocierade molekylära mönster uttryckta av patogener och mönsterigenkänningsreceptorer uttryckta av värdceller vid cellytan (avgiftsliknande receptorer och C-typ lektinreceptorer), i endosomen (TLR), eller i cytoplasman (retinsyrainducerbar gen 1-liknande receptorer och nukleotidbindande oligomeriseringsdomänliknande receptorer).
Konsekvensen av överdriven inflammation är säkerhetsvävnadsskador och nekrotisk celldöd, vilket resulterar i frisättning av skadeassocierade molekylära mönster, så kallade faramolekyler som upprätthåller inflammation åtminstone delvis genom att agera på samma mönsterigenkänningsreceptorer som utlöses av patogener.