Angelman syndrom (AS) er en sjælden neurogenetisk lidelse, der rammer ca. en ud af 15.000 mennesker – ca. 500.000 individer verden over. Børn og voksne med AS har typisk balanceproblemer, motorisk svækkelse og svækkende anfald. Nogle mennesker går aldrig. De fleste taler ikke. Forstyrrede søvncyklusser kan også være en alvorlig udfordring for den enkelte og vicevært. Personer med AS kræver kontinuerlig pleje og er ikke i stand til at leve uafhængigt. De har en normal forventet levetid. Dette er livet i dag for mennesker, der lever med Angelman syndrom, men håbet er her. Forskere mener, at AS har det største potentiale for at blive helbredt sammenlignet med andre neurogenetiske lidelser, og FAST (Foundation for Angelman Syndrome Therapeutics) har en plan godt i gang for at opnå netop det.
Typer Årsager Tests & Diagnoseressourcer
Typer af Angelman-syndrom
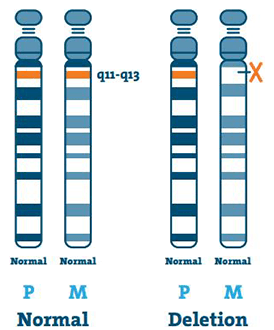
Sletning ( 65-75%)
DNA (deoxyribonukleinsyre) er hovedkomponenten i kromosomer. Den indeholder vores unikke genetiske kode. De fleste individer med AS mangler et stykke DNA i region 15q11-13 på moderens kromosom 15.
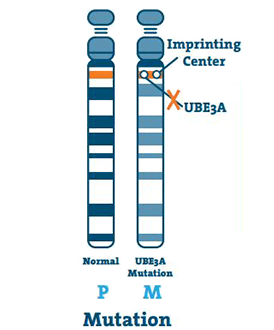
Mutation (5 -11%)
Dette sker, når der er en lille abnormitet i DNA’et i UBE3A-genet. En mutation kan ske hvor som helst på genet.
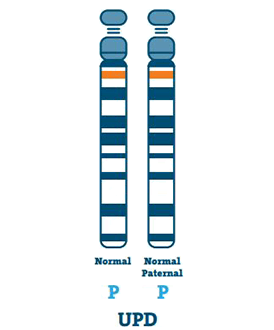
Uniparental Disomy (3-7%)
En person med UPD har to kopier af kromosom 15 fra deres far i stedet for en hver fra far og mor. UPD sker normalt, hvis der ikke er noget kromosom 15 i ægget.
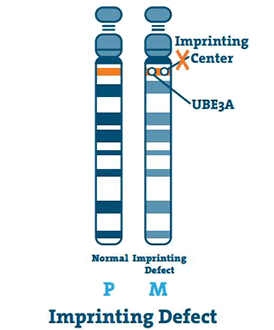
Imprinting Defect (< 3%)
Aftrykningscentret er en lille strækning af DNA placeret i kromosomets q11-13-region. I sjældne tilfælde er moderens kromosom 15 tom, og centret kopierer farens kromosom 15. Dette er en imprintingsfejl.
Karakteristika
Typiske egenskaber ved AS er normalt ikke tydelige ved fødsel. Mennesker med lidelsen har fodringsproblemer som spædbørn og mærkbar forsinket udvikling omkring 6-12 måneder. De har brug for intensive terapier for at hjælpe med at udvikle funktionelle færdigheder. AS påvirker hvert løb og begge køn. Det diagnosticeres ofte fejlagtigt som autisme eller cerebral parese.
Adfærdsmæssige træk
Mennesker med Angelman-syndrom har nogle adskilte adfærdstræk, herunder en glad opførsel, der er karakteriseret ved hyppig latter, smil og ophidselse. Mange personer med AS har en fascination af vand og nyder stor glæde i aktiviteter som svømning og badning.
Årsager til Angelmans syndrom
Angelman syndrom er en enkelt-gen-lidelse forårsaget af et tab af funktion i UBE3A-genet på moderens 15. kromosom. Folk har to sæt kromosomer – et arvet fra moderen og et fra faren. I en typisk person er det maternelt nedarvede UBE3A-gen aktivt, mens kopien af genet arvet fra faderen tavs i neuronerne i vores hjerner – et fænomen kendt som imprinting. For mennesker med AS gør dette moderlige gen ikke sit job, og det påvirker deres Messenger-RNA (mRNA).
![]()
Hvad er mRNA?
mRNA er kroppens FedEx. Vores DNA bruger mRNA som en leveringstjeneste til at sende tegninger til proteinsamlingsfabrikkerne i vores celler. Mennesker med AS har en mutation, sletning eller anden defekt i deres UBE3A-gen, der afbryder denne leveringstjeneste. Som et resultat fremstiller deres neuroner ikke noget funktionelt UBE3A-protein, og det er det, der udløser symptomerne på AS. Dette protein er det, der hjælper os med at gå, tale og udføre andre daglige opgaver.
![]()
Er det genetisk?
I de fleste tilfælde arves ikke Angelman syndrom – især dem, der er forårsaget af en sletning eller UPD. I stedet for forekommer disse genetiske ændringer som tilfældige hændelser under dannelsen af reproduktive celler eller i den tidlige embryonale udvikling.
Test & Diagnose
Lær om de kriterier, som læger bruger til at bestemme, om et individ har Angelman-syndrom, og læse om tilgængelige tests og almindelige fejldiagnoser.
![]()
Test
![]()
Diagnose

Deltag i vores fællesskab
Opret forbindelse til andre mennesker, der har et barn eller elsket en med Angelman-syndrom.
