Introduktion
I årtier har mad-lægemiddelinteraktioner (FDI) og urt-lægemiddelinteraktioner været kendt for at begrænse succesen med medicinske behandlinger. Det enorme antal mulige interaktioner mellem genetiske variationer, medicinske regimer og de mange bioaktive forbindelser, der findes i mad og urter, resulterer i overvældende kompleksitet. Moderne værktøjer som big-data-analyse, maskinindlæring og simulering af protein-ligand-interaktioner kan hjælpe os med at besvare et helt sæt spørgsmål: Kan madvalg bidrage til, at terapeutiske regimer mislykkes, og i bekræftende fald hvordan? Hvilke (n) mad (er) skal indtages før du tager et ordineret lægemiddel? Og sandsynligvis det mest spændende spørgsmål: Hvordan kan vi bruge disse værktøjer til at forudsige personlig FDI? Det er klart, at mange svar ligger i stofskiftet af stoffer, fødevarer og urter fra cytochrom P450 3A4 (CYP3A4) i leveren og fordøjelseskanalen (Galetin et al., 2010; Basheer og Kerem, 2015).
De fleste gener, der koder for CYP-enzymer, er polymorfe. Til dato er den mest omfattende informationskilde, der beskriver CYP-alleler, Pharmacogen Variation Consortium1, hvor færre end 100 alleler af CYP3A4 er repræsenteret. Af disse er færre end 40 exoniske SNP’er (enkelt nukleotidpolymorfier), der resulterer i en modificeret proteinsekvens. Det lille antal forsøgspersoner i alle tidligere offentliggjorte værker om CYP3A4-mutationer giver os begrænsede data vedrørende sande frekvenser af CYP3A4-mutationer i hele befolkningen og i definerede grupper.
Ikke kun at pålidelig information om SNPs-forekomst er ufuldstændig , også er deres kliniske implikationer endnu uklare i de fleste tilfælde (Zanger et al., 2014). At forstå hvilke og hvornår SNP’er kan have klinisk betydning er en enormt kompleks opgave. In vitro-analyser er tidskrævende, dyre og praktisk talt af lav relevans i betragtning af den store mængde mutationer og det uendelige antal mad-lægemiddelkombinationer. Molekylære modelleringsmetoder, herunder docking og fri energi bindende beregninger, kan tjene til at forudsige potentielle effekter af SNP’er og af mange forbindelser på CYP3A4-medieret stofskifte (Lewis et al., 1998). For eksempel bidrager ikke-kovalente, hydrofobe, elektrostatiske og van der Waals-vekselvirkninger alle til orienteringen af en forbindelse og dermed til dens binding og reaktion på et enzyms aktive sted. Til gengæld vil disse bestemme enzymets affinitet og specificitet for forskellige substrater og styrken af enzymhæmmere (Kirchmair et al., 2012; Basheer et al., 2017).
Her foreslår vi en ny tilgang til måling af allelfrekvensen af CYP3A4-mutationer i forskellige etniske grupper. Denne omfattende tilgang har beføjelse til at fremhæve mutationer, der er udbredte i bestemte etniske grupper, og kombineret med screening for interagerende kemikalier, f.eks. Hæmmere fra mad, tillader belysning af virkningerne af bestemte mutationer på lægemiddel-mad-interaktion, der tjener som en indledende skridt mod personlig medicin og ernæring. Dette arbejde kan øge bevidstheden om den mulige kliniske betydning af proteinændrende CYP3A4 SNP’er og foreslår også et par nødvendige værktøjer til fremme og anvendelse af præcision og personlig medicin.
Materialer og metoder
Databasescreening og dataanalyse
CYP3A4-varianter datasættet blev downloadet fra gnomAD-browseren2 som en CVS-fil. Python 2.7 med NumPy, pandaer og matplotlib-pakker blev brugt til dataanalyse og visualisering (se Supplerende datablad S1). Agglomerativ hierarkisk klyngedannelse blev udført ved hjælp af Expander 7-softwaren (Shamir et al., 2005) med Pearson rang-korrelationskoefficienten som et mål for ligheder og komplet koblingstype. En afstandstærskel på 0,6 blev indstillet til gruppering af SNP’er.
I silico blev Polymorphism Modelling
Maestro 2017-2 frigivelse (Schrodinger, New York, NY, USA) brugt til beregningsmodellering. CYP3A4-dockingmodel blev bygget som tidligere beskrevet (Basheer et al., 2017). Kort fortalt blev CYP3A4-krystalstruktur (PDB-indgang 2V0M) behandlet, modificeret og raffineret efter trinene i guiden til proteinforberedelse. Et docking-gitter med en metal-koordineringsbegrænsning for Fe2 + i hæmgruppen blev genereret baseret på centroid af ketoconazol i det oprindelige bindingssted i krystalstrukturen. Syv mutationer blev valgt til docking-simuleringer, en som repræsentant for hver etnisk gruppe (tabel 1, 2). For hvert variantprotein blev en enkeltpunktsmutation indført før proteinfremstillingstrin. 3D-strukturer af ligander blev genereret baseret på 2D-strukturer fra PubChem3 og forberedt til docking ved hjælp af LigPrep-opgave.OPLS3 kraftfelt og standardglideindstillinger for standardpræcision blev anvendt til dockingmodellen med den undtagelse, at metalkoordinationsbegrænsningen blev brugt samt 30 stillinger for antallet af stillinger, der skal inkluderes, og 10 stillinger for antallet af stillinger, der skal skrives ud. For hver ligand blev dockingsresultatet med den laveste Glide-emodel score valgt.

Tabel 1. Udvalgte repræsentative SNP’er for syv etniske grupper.

Tabel 2. Frekvens (%) af valgte mutationer efter etnisk gruppe.
Resultater
Genomaggregationsdatabasen (gnomAD; se tekstfodnote 2) samler både exome- og genom-sekventeringsdata fra en lang række store sekvenseringsprojekter. Det inkluderer data fra 125.748 eksomsekvenser og 15.708 helgenomsekvenser fra 141.456 ikke-relaterede individer, der repræsenterer syv etniske populationer (Lek et al., 2016). GnomAD-databasen præsenterer 856 varianter af CYP3A4, hvoraf 397 er introniske og så mange som 459 exonic. Af de eksoniske SNP’er er 312 missense-mutationer, hvilket indikerer, at de påvirker proteinstrukturen. CYP3A4-genet er 34.205 bp langt. Dens 13 eksoner omfatter en 1,512-bp kodende region, der producerer et protein med 504 aminosyrer. De 412 exoniske SNP’er med unikke positioner i dette gen resulterer i en exonisk SNP-tæthed på 272 / kbp (supplerende tabel S1).
Beregning af differentierede allelfrekvenser pr. Etnisk gruppe afslører, at nogle populationer udviser højere mutationsfrekvenser (Figur 1A). De fleste af CYP3A4-mutationerne i den europæiske befolkning er faktisk sjældne, som det almindeligvis antages, mens mutationer i andre populationer, såsom afrikanske og østasiatiske, er meget mere udbredte (supplerende tabel S2).
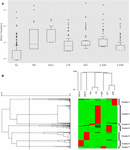
Figur 1. Analyse af CYP3A4 missense SNP’er i syv forskellige populationer. (A) Log-skala boks plot af allel frekvenser. Kasser repræsenterer interkvartilområdet (IQR), blå linjer repræsenterer medianerne, whiskers repræsenterer data inden for 1,5 IQR, og outliers vises som små cirkler. (B) Hierarkisk gruppering af allelfrekvenser. Hver række repræsenterer en enkelt SNP. Hver kolonne repræsenterer særskilt etnisk befolkning. SNPernes allelfrekvens i hver af populationerne er repræsenteret af farven på den tilsvarende celle i matrixfilen. Grøn og rød repræsenterer henholdsvis lav og høj frekvens. Det øverste dendrogram viser ligheder i allelfrekvensmønsteret mellem hver gruppe af forsøgspersoner. Venstre dendrogram repræsenterer gruppering af gener i to grupper. Den stiplede linje repræsenterer 0,6 afstandstærsklen, der bruges til opdeling til grupper. EU – europæisk (ikke-finsk; n = 64.603), FIN – europæisk (finsk; n = 12.562), ASH J – Ashkenazi-jødisk (n = 5.185), LTN – latino (n = 17.720), AFR – afrikansk (n = 12.487), E ASN – østasiatiske (n = 9.977), S ASN – sydasiatiske (n = 64.603).
Vi brugte hierarkisk klyngedannelse til at gruppere varianter med lignende frekvensmønstre. Vores dataanalyse gav syv forskellige klynger (figur 1B). Desuden observeres det tydeligt, at højfrekvente SNP’er i hver klynge er karakteristiske for en specifik population. Hierarkisk klyngeanalyse af de etniske grupper understøtter sammenhængen mellem genetisk varians og etnicitet ved at gruppere beslægtede etniske grupper som syd- og østasiere såvel som finske og ikke-finske europæere.
En beregningsmodel blev brugt til at vurdere den mulige indflydelse af punktmutationer i CYP3A4 på dets evne til at binde substrater og hæmmere. CYP3A4 er i stand til at oxidere en bred vifte af endogene og xenobiotiske forbindelser. Her blev ketoconazol valgt som et repræsentativt lægemiddel og en meget effektiv specifik inhibitor; androstenedion og testosteron blev valgt som repræsentativt endogent hormon; og demethoxycurcumin og epigallocatechin blev valgt som repræsentanter for diætbioaktive stoffer. En docking-model blev bygget til at forudsige bindingspositionerne for de valgte forbindelser i CYP3A4-bindingsstedet. Modellen blev først valideret ved succesfuld gendannelse af ketoconazolposen i bindingsstedet med en RMSD på 1,52 Å i forhold til den oprindelige krystalstruktur. Syv mutante proteiner blev designet baseret på krystalstrukturen af vildtypeproteinet (supplerende figur S1). For hver etnisk gruppe blev den hyppigste unikke mutation valgt som repræsentativ. Virkningen af enkeltmutationer på substratbinding blev vurderet baseret på sammenligningen mellem docking udgør på det native protein og på variantproteiner. Ændringer i dockingstillinger med hensyn til RMSD er opsummeret i tabel 3.

Tabel 3. RMSD i forhold til WT for dockingligander til bindingssteder i syv CYP3A4-varianter.
Virkningen af CYP3A4 SNP på substratbinding viste sig at være mutationssubstratspecifik. Kun i nogle få tilfælde forårsagede mutationer en ændring i bindingspositionen for en ligand i bindingslommen. Testosteron-docking udgør den samme i alle syv testede varianter. E262K-, D174H- og K168N-varianterne forårsagede ikke en binding udgør ændring i nogen af de testede molekyler. L373F- og T163A-mutationerne ændrede imidlertid androstenedionens bindingsposition, så den blev positioneret parallelt med hemgruppen snarere end vinkelret på den, som i WT-proteinet. Androstenedion blev også roteret således, at cyclopentanon-gruppen er placeret proximalt til hæm i stedet for cyclohexanon-gruppen i WT-proteinet. S222P- og L293P-mutationerne forårsagede kun en lille rotation i bindingspositionen af androstenedion (figur 2A). Af alle undersøgte mutationer forårsagede kun S222P væsentlige ændringer i dockingposer af ketoconazol og demethoxycurcumin på bindingsstedet (figur 2B, C); derimod for epigallocatechin var den positureskiftende mutation L373F (figur 2D).
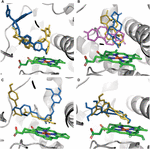
Figur 2. Modeller af ligander forankret ved bindingsstedet for CYP3A4. (A) Ketoconazol, (B) androstenedion, (C) demethoxycurcumin og (D) epigallocatechin. Det proteinbindende sted er repræsenteret af grå bånd; heme er repræsenteret af grønne pinde, docking udgør i WT-proteinet, og i S222P og L373F er mutanter vist som henholdsvis orange, blå og violette pinde. Androstenedione docking udgør i L293P og i T136A varianter overlapper stillingerne i henholdsvis S222P og L373F varianter.
Diskussion
Cytochrom P450 3A4 er det vigtigste enzym, der er ansvarlig for mad-lægemiddelinteraktioner. Nuværende forskning i mutationer i CYP3A4 har været fokuseret på et par dusin SNP’er fundet i udpegede studier (Sata et al., 2000; Dai et al., 2001; Eiselt et al., 2001; Hsieh et al., 2001; Lamba et al. ., 2002; Murayama et al., 2002). Som vist her repræsenterer de toppen af et isbjerg i betragtning af prævalensen og de potentielle resultater af CYP3A4-mutationer. Overfloden af store genom- og exome-sekventeringsprojekter har åbnet en ny vej til identifikation af mange ukendte mutationer. Her viser vi, at de tidligere præsenterede mutationer kun er toppen af isbjerget ved at demonstrere 856 mutationer, der findes i CYP3A4, hvoraf en tredjedel ændrer proteinstrukturen. Ved hjælp af en kohorte på 141.456 ikke-relaterede individer blev nøjagtige allelfrekvenser af CYP3A4-mutationer beregnet for syv separate etniciteter. Så vidt vi ved, er dette den største og mest omfattende store datastudie af CYP3A4 exoniske mutationer og deres allelfrekvenser i forskellige populationer, offentliggjort til dato.
Polymorfe CYP3A4 enzymer kan være meget vigtige i forklaringen forskelle i lægemiddeleffektivitet og toksicitet blandt forskellige individer. Mutationer i CYP3A4-genet kan føre til afskaffet, reduceret, ændret eller øget enzymatisk aktivitet. Eksoniske mutationer kan ændre enzymatisk aktivitet, som det er blevet demonstreret i nogle få kliniske studier med udvalgte substrater. Nogle tilfælde af ændret stofskifte på grund af SNP’er i CYP3A4 er allerede beskrevet i litteraturen (Eiselt et al., 2001; Miyazaki et al., 2008). På trods af SNP’ers funktionelle betydning og kliniske relevans i CYP3A4 og muligvis på grund af deres relativt lave identificerede hyppighed i den generelle befolkning, har polymorfisme i CYP3A4 ikke fået den opmærksomhed, det fortjener.
Her tjente syv mutationer til at forudsige effekten af SNP’er på substrat- og inhibitorbindingsorientering. I litteraturen opdeler CYP3A4 polymorfisme den generelle befolkning i tre grupper – dårlige metaboliserere, normale metaboliserere og hurtige metaboliserere, baseret på introniske SNP’er, der ændrer ekspressionsniveauer snarere end struktur (Zanger og Schwab, 2013). Vores beregninger antyder en yderligere klassificering: de ændrede metaboliserere. Nogle mutationer foreslået af vores virtuelle model ville medføre en ændring i bindingsorienteringen af individuelle ligander. Disse ændringer forventes at mindske sandsynligheden for enzymatisk oxidation på grund af øget afstand fra hæmmen eller føre til produkter, der ellers ikke ville være tydelige under toksicitetstest udført som en del af lægemiddeludviklingsprocessen. Som vores model forudsiger, er CYP3A4-mutationer for de fleste substrater godartede.
Modificeret position af et substrat i bindingslommen på grund af protein strukturændring er kun en mulig mekanisme, hvormed en mutation kan ændre et proteins aktivitet . Nedsat forankring af proteinet til membranen, beskadigede substratledende kanaler og kompromitteret udgang af produkterne giver yderligere mekanismer til en mutationsændring i et proteins aktivitet. Som vist her er effekten af enhver mutation substratspecifik.Det er besværligt at bestemme, hvilke kombinationer af substrater og mutationer der kan ændre den enzymatiske aktivitet ved hjælp af traditionelle in vitro-metoder, idet det understreges behovet for forudsigelige virtuelle værktøjer til løsning af dette komplekse puslespil.
Offentlig og professionel interesse i personlig medicin og præcisionsmedicin vokser hurtigt. Forudsigelse af modificeret lægemiddelmetabolisme baseret på individuel polymorfisme i CYP3A4 synes kun at være et spørgsmål om tid. Her foreslår vi, at forskellige etniske grupper bærer unikke sæt CYP3A4 SNP’er. Faktisk kan etnicitet tjene som et første gennemførligt skridt inden for personlig medicin, før implementeringen af en individuel DNA-skærm for alle. Interessant nok har etnicitet en yderligere implikation for CYP3A4-stofskifte, idet den er en vigtig faktor i bestemmelsen af madvalg og diætvaner. Det kan foreslås, at terapeutiske regimer skal være specifikt designet til hver etnisk gruppe, i det mindste for lægemidler, der er stærkt metaboliseret af CYP3A4. Dette fremhæver mulighederne for at udnytte og integrere databaser og dyb læring for at identificere, hvordan SNP’er, etnicitet, diætforbindelser og stoffer ændrer CYP3A4-aktivitet og succesen med et medicinsk regime.
Datatilgængelighed
Offentligt tilgængelige datasæt blev analyseret i denne undersøgelse. Disse data kan findes her: http://gnomad.broadinstitute.org/gene/ENSG00000160868.
Forfatterbidrag
Alle nævnte forfattere har ydet et betydeligt, direkte og intellektuelt bidrag til arbejde og godkendte det til offentliggørelse.
Erklæring om interessekonflikt
Forfatterne erklærer, at forskningen blev udført i mangel af kommercielle eller økonomiske forhold, der kunne opfattes som et potentiale interessekonflikt.
Supplerende materiale
FIGUR S1 | 3D-båndmodel af CYP3A4 og placeringen af de muterede aminosyrer i de syv variantproteiner designet til docking. Heme er repræsenteret som grønne pinde, Fe2 + er repræsenteret som en rød kugle, SNP’er anvendt i in silico-analyse er repræsenteret som røde områder på båndet, og R-grupper af muterede aminosyrer i variantmodeller vises eksplicit som lysegrå pinde.
TABEL S1 | CYP3A4 SNP-typer i en befolkning på 141, 456 ikke-relaterede personer, der repræsenterer 7 etniske befolkninger.
TABEL S2 | CYP3A4 SNP’er efter etnisk gruppe.
Fodnoter
- ^ www.pharmvar.org
- ^ https://gnomad.broadinstitute.org
- ^ https://pubchem.ncbi.nlm.nih.gov