Malign intravaskulær betændelse
Sepsis er blevet omtalt som en proces med ondartet intravaskulær inflammation . Normalt sikrer en potent, kompleks, immunologisk kaskade en hurtig beskyttende reaktion på mikroorganismes invasion hos mennesker. Et mangelfuldt immunologisk forsvar kan muliggøre, at infektion etableres; dog kan en overdreven eller dårligt reguleret reaktion skade værten gennem maladaptiv frigivelse af oprindeligt genererede inflammatoriske forbindelser (se billedet nedenfor).
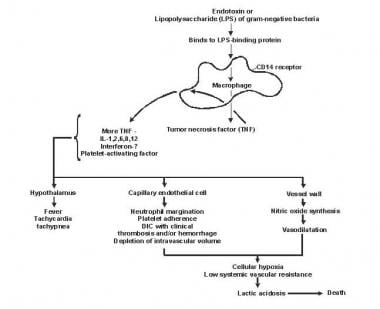 Patogenese af sepsis og multiorgansvigt.
Patogenese af sepsis og multiorgansvigt. Lipid A og andre bakterielle produkter frigiver cytokiner og andre immunmodulatorer, der medierer de kliniske manifestationer af sepsis. Interleukiner, tumornekrosefaktor (TNF) -α, interferon gamma (IFN-γ) og andre kolonistimulerende faktorer produceres hurtigt inden for få minutter eller timer efter interaktioner mellem monocytter og makrofager med lipid A.
Inflammatorisk mediatorfrigivelse bliver en selvstimulerende proces og frigivelse af andre sådanne mediatorer, herunder interleukin (IL) -1, blodpladeaktiverende faktor, IL-2, IL-6, IL-8, IL-10 og nitrogenoxid (NO) øger cytokinniveauet yderligere. Dette fører til fortsat aktivering af polymorfonukleære leukocytter (PMN’er), makrofager og lymfocytter; proinflammatoriske mediatorer rekrutterer flere af disse celler. Alle disse processer skaber en tilstand af destruktiv immunologisk dissonans.
Sepsis er beskrevet som en autodestruktiv proces, der tillader udvidelse af det normale patofysiologiske respons på infektion for at involvere ellers normalt væv og resultater i MODS. Organdysfunktion eller organsvigt kan være det første kliniske tegn på sepsis, og intet organsystem er immun over for konsekvenserne af de inflammatoriske overdreven sepsis. Dødeligheden stiger, når organsvigt stiger.
Selvom ukontrolleret, når MODS først udvikler systemisk bevis på både proinflammatorisk og antiinflammatorisk opregulering, er der normalt til stede, hvilket tyder på, at fiasko ved værtsforsvars homeostase er den sidste vej fra sepsis til MODS snarere end simpel hypotension-induceret end-organskade, som det kan forekomme med hæmoragisk chok. Overlevelse fra svær sepsis med MODS er normalt forbundet med en generaliseret reduktion i både det proinflammatoriske og antiinflammatoriske respons.
En ny hypotese er for nylig opstået, at overlevelse fra svær sepsis kræver en generaliseret nedregulering af kroppens immunrespons, energiske funktioner og tilhørende organydelse. Således kan MODS ved værtens adaptive reaktion på overvældende betændelse, hvilket tillader betændelse at rydde uden at forårsage permanent skade på slutorganet. Som diskuteret nedenfor afslører alle organer en generaliseret hyporesponsivitet, der klart er unormal i helbredet, men som kan markere en overlevelsesstrategi ved svær sepsis.
Dysfunktion i organsystemer
Cirkulationsforstyrrelse
Væsentlig forstyrrelse i autoregulering af cirkulation er typisk for sepsis. Vasoaktive mediatorer forårsager vasodilatation og øger mikrovaskulær permeabilitet på infektionsstedet. NO spiller en central rolle i vasodilatationen af septisk chok. Desuden kan nedsat sekretion af vasopressin forekomme, hvilket kan muliggøre vedvarende vasodilatation.
Ændringer i både systolisk og diastolisk ventrikulær ydeevne forekommer i sepsis. Ved brug af Frank-Starling-mekanismen øges hjertevolumen ofte for at opretholde blodtrykket i nærvær af systemisk vasodilatation. Patienter med allerede eksisterende hjertesygdomme er ude af stand til at øge deres hjerteoutput korrekt.
Regionalt interfererer sepsis med den normale fordeling af systemisk blodgennemstrømning til organsystemer. Derfor kan kerneorganer muligvis ikke modtage passende iltafgivelse, og resultatet er det, der kaldes regional hypoperfusion.
Mikrocirkulation er det vigtigste målorgan for skade ved sepsis, da vaskulært endotel er universelt påvirket af de cirkulerende inflammatoriske mediatorer. Selv om det er uklart, om mikrocirkulationsdannelsesforstyrrelser er årsagen eller en uskyldig tilskuer til endorganets skade, ses en klar mikrovaskulær dysfunktion. Der ses et fald i antallet af perfunderede kapillærer, skønt der ved anvendelse af vasodilaterende terapier forekommer fuld mikrovaskulær rekruttering. Mitokondrie dysfunktion forekommer også og er ofte forbundet med reducerede mitokondrie transmembrane potentialgradienter, som er nødvendige for at drive oxidativ fosforylering. Slutresultatet er en tilsyneladende manglende evne hos slutorganer til at udvinde ilt maksimalt.
Debatten fortsætter om, hvorvidt denne svigt i energimetabolisme er en adaptiv cytobeskyttende mekanisme, der ligner dvale eller afspejler primær mitokondriepatologi. Dette er områder med aktiv forskning, men oversættes ikke i øjeblikket til klare retningslinjer for klinisk praksis. Øget kapillær endotelpermeabilitet fører til udbredt proteinrig vævsødem.
Septisk chok og SIRS er kendetegnet ved reversibel myokardie-depression, som kan vise sig at være resistent over for administration af catecholamin og væske. Cirkulerende “myokardial depressiv faktor” – sandsynligvis repræsenterende de synergistiske virkninger af TNF-α, IL-1β, andre cytokiner og NO – er impliceret i patogenesen. De to karakteristika ved denne akutte stress-myokardie-depression er nedsat adrenerg respons og diastolisk dysfunktion, der fører til relativ katecholaminresistens og små snarere end dilaterede hjerter Makrovaskulær myokardieiskæmi og hypoperfusion er usandsynlige bidragydere.
I svær sepsis og septisk chok forårsager mikrocirkulationsdysfunktion og mitokondrie depression regional vævsnød, og regional dysoxi vedvarer derfor. Denne tilstand betegnes mikrocirkulations- og mitokondrie-distress-syndrom (MMDS). Sepsis-induceret inflammatorisk autoregulatorisk dysfunktion vedvarer, og iltbehovet matches ikke med forsyning, hvilket fører til MODS.
Omfordeling af intravaskulært væskevolumen som følge af nedsat arteriel vaskulær tone, nedsat venøs tilbagevenden fra venøs di lation og frigivelse af myokardiedepressive stoffer forårsager hypotension.
Pulmonal dysfunktion
Endotelskade i lungevaskulaturen fører til forstyrret kapillærblodgennemstrømning og forbedret mikrovaskulær permeabilitet, hvilket resulterer i interstitielt og alveolært ødem. Neutrofil indeslutning i den pulmonale mikrocirkulation initierer og forstærker skaden på alveolære kapillarmembraner. Akut lungeskade og akut åndedrætssyndrom (ARDS) er hyppige manifestationer af disse effekter. Faktisk er sepsis og lungebetændelse de mest almindelige årsager til ARDS.
Gastrointestinal dysfunktion
Mave-tarmkanalen (GI) kan hjælpe med at udbrede skaden af sepsis. Overvækst af bakterier i den øvre mave-tarmkanal kan opsuges i lungerne, hvilket producerer nosokomial eller aspirationspneumoni. Tarmens normale barrierefunktion kan blive påvirket, hvilket tillader translokation af bakterier, endotoksiner og normale fordøjelsesproteaser i den systemiske cirkulation og forlænger septisk respons.
Septisk shock kan forårsage paralytisk ileus, der kan føre til en forsinkelse i institutionen for enteral fodring. Overskydende NO-produktion menes at være det forårsagende middel til sepsis-induceret ileus. Det optimale niveau af ernæringsindtag forstyrres i lyset af høje protein- og kaloribehov. Narkotika og muskelafslappende midler kan yderligere forværre mave-tarmkanalen.
Leverdysfunktion
Som en konsekvens af den rolle, leveren spiller i værtsforsvar kan de unormale syntetiske funktioner forårsaget af leverdysfunktion bidrage til både initiering og progression af sepsis. Det retikuloendoteliale system i leveren fungerer som en første forsvarslinje til at rydde bakterier og deres produkter; leverdysfunktion fører til et overskud af disse produkter til systemisk cirkulation.
Leversvigt (“chokeret lever”) kan manifestere sig ved forhøjelser i leverenzymer og bilirubin, koagulationsdefekter og manglende udskillelse af toksiner såsom ammoniak, som fører til forværret encefalopati.
Nyredysfunktion
Akut nyreskade (AKI) ofte ledsaget sepsis. Forskellige etiologier for AKI er blevet rapporteret, og årsagen antages typisk at være multifaktoriel. AKI-mekanismen er kompleks, men involverer sandsynligvis et fald i effektivt intravaskulært volumen som følge af systemisk hypotension, direkte renal vasokonstriktion, frigivelse af cytokiner, og aktivering af neutrofiler af endotoksiner og andre peptider, som bidrager til nyreskade. Stadig viser de fleste dyreforsøg, at renal blodgennemstrømning øges, ikke nedsættes, i sepsis, selvom det er forbundet med nedsat rørformet funktion og mangel på signifikant histologisk bevis for tu bular skade.
Dysfunktion i centralnervesystemet
Inddragelse af centralnervesystemet (CNS) i sepsis producerer encefalopati og perifer neuropati. Patogenesen er dårligt defineret, men er sandsynligvis relateret til systemisk hypotension, hvilket kan føre til hjernens hypoperfusion.
Koagulopati
Subklinisk koagulopati, signaleret ved en mild forhøjelse af trombintiden (TT) eller aktiveret delvis tromboplastintid (aPTT) eller en moderat reduktion i blodpladetallet er ekstremt almindeligt; dog kan åbenlyst spredt intravaskulær koagulation (DIC) også udvikle sig. Protease-aktiverede receptorer (PAR’er), især PAR 1, danner den molekylære forbindelse mellem koagulation og inflammation; PAR1 udøver cytobeskyttende effekter, når de stimuleres af aktiveret protein C eller lavdosis thrombin, men udøver forstyrrende virkninger på endotelcellebarrierefunktionen, når de aktiveres af højdosis thrombin.
Mekanismer for organdysfunktion og -skade
De nøjagtige mekanismer for celleskade og resulterende organdysfunktion i sepsis forstås ikke fuldt ud. MODS er associeret med udbredt endotel- og parenkymcellebeskadigelse, hvoraf nogle kan forklares med følgende 4 foreslåede mekanismer.
Hypoxisk hypoxi
Septisk kredsløbslæsion forstyrrer iltning af væv, ændrer den metaboliske regulering af vævsiltlevering og bidrager til organdysfunktion. Mikrovaskulære og endotel abnormiteter bidrager til septisk mikrocirkulationsdefekt i sepsis. De reaktive iltarter, lytiske enzymer og vasoaktive stoffer (f.eks. NO- og endotelvækstfaktorer) fører til mikrocirkulationsskade, hvilket er forstærket af erytrocyternes manglende evne til at navigere i septisk mikrocirkulation. >
Direkte cytotoksicitet
Endotoksin, TNF-α og NO kan forårsage skade på mitokondrie elektrontransport, hvilket fører til forstyrret energimetabolisme. Dette kaldes cytopatisk eller histotoksisk anoxi, en manglende evne til at bruge ilt, selv når den er til stede.
Apoptose
Apoptosis ( programmeret celledød) er den vigtigste mekanisme, hvormed dysfunktionelle celler normalt elimineres. De proinflammatoriske cytokiner kan forsinke apoptose i aktiverede makrofager og neutrofiler, men andre væv (f.eks. Tarmepitel) kan gennemgå accelereret apoptose. Derfor spiller forstyrrelse af apoptose en kritisk rolle i vævsskade ved sepsis.
Immunosuppression
Interaktionen mellem proinflammatorisk og antiinflammatoriske mediatorer kan føre til ubalance mellem dem. En inflammatorisk reaktion eller en immundefekt kan være dominerende, eller begge kan være til stede.
Værtsrespons og andre faktorer, der påvirker resultatet
Kliniske egenskaber, der relaterer til sværhedsgraden af sepsis inkluderer værtsrespons på infektion, stedet og typen af infektion, timingen og typen af antimikrobiel terapi, den krænkende organisme, udviklingen af chok, den underliggende sygdom, patientens langsigtede helbredstilstand og antallet af mislykkede organer. Faktorer, der fører til sepsis og septisk chok, er muligvis ikke nødvendige for at bestemme det ultimative resultat.
Værtsresponsen mod sepsis er kendetegnet ved både proinflammatoriske responser og antiinflammatoriske immunsuppressive responser. Retningen, omfanget og varigheden af disse reaktioner bestemmes af både værtsfaktorer (f.eks. Genetiske egenskaber, alder, sameksisterende sygdomme, medicin) og patogenfaktorer (fx mikrobiel belastning, virulens).
Inflammatoriske reaktioner initieres ved interaktion mellem patogenassocierede molekylære mønstre udtrykt af patogener og mønstergenkendelsesreceptorer udtrykt af værtsceller på celleoverfladen (toll-lignende receptorer og C-type lectinreceptorer), i endosomet (TLR’er) eller i cytoplasmaet (retinsyre-inducerbart gen 1-lignende receptorer og nukleotidbindende oligomeriseringsdomæne-lignende receptorer).
Konsekvensen af overdreven betændelse er sikkerhedsskader på væv og nekrotisk celledød, hvilket resulterer i frigivelse af skaderelaterede molekylære mønstre, såkaldte faremolekyler, der i det mindste opretholder betændelse delvis ved at virke på de samme mønstergenkendelsesreceptorer udløst af patogener.