Introducción
Durante décadas, se ha sabido que las interacciones entre alimentos y medicamentos (IED) y las interacciones entre hierbas y medicamentos limitan el éxito de los tratamientos médicos. La enorme cantidad de posibles interacciones entre las variaciones genéticas, los regímenes médicos y los numerosos compuestos bioactivos que se encuentran en los alimentos y las hierbas resultan en una complejidad abrumadora. Las herramientas modernas como el análisis de macrodatos, el aprendizaje automático y la simulación de interacciones proteína-ligando pueden ayudarnos a responder una serie de preguntas: ¿Podrían las elecciones alimentarias contribuir al fracaso de los regímenes terapéuticos? ¿Qué alimentos se deben consumir antes de tomar un medicamento recetado? Y probablemente la pregunta más interesante: ¿cómo podemos utilizar estas herramientas para predecir la IED personal? Claramente, muchas respuestas se encuentran en el metabolismo de medicamentos, alimentos y hierbas por el citocromo P450 3A4 (CYP3A4) en el hígado y el tracto digestivo (Galetin et al., 2010; Basheer y Kerem, 2015).
La mayoría de los genes que codifican las enzimas CYP son polimórficos. Hasta la fecha, la fuente de información más completa que detalla los alelos de CYP es el Consorcio de Variaciones Farmacogénicas1, en el que están representados menos de 100 alelos de CYP3A4. De estos, menos de 40 son SNP exónicos (polimorfismos de un solo nucleótido) que dan como resultado una secuencia de proteína modificada. El pequeño número de sujetos en todos los trabajos publicados anteriormente sobre las mutaciones del CYP3A4 nos proporciona datos limitados con respecto a las verdaderas frecuencias de las mutaciones del CYP3A4 en toda la población y en grupos definidos.
No solo la información confiable sobre la incidencia de SNP es incompleta. , además, sus implicaciones clínicas aún no están claras en la mayoría de los casos (Zanger et al., 2014). Entender cuáles y cuándo los SNP pueden tener importancia clínica es una tarea tremendamente compleja. Los ensayos in vitro requieren mucho tiempo, son costosos y prácticamente de poca relevancia considerando la gran cantidad de mutaciones y el número infinito de combinaciones de alimentos y medicamentos. Los métodos de modelado molecular, incluidos los cálculos de acoplamiento y unión de energía libre, pueden servir para predecir los efectos potenciales de los SNP y de muchos compuestos sobre el metabolismo mediado por CYP3A4 (Lewis et al., 1998). Por ejemplo, las interacciones no covalentes, hidrófobas, electrostáticas y de van der Waals contribuyen a la orientación de un compuesto y, por lo tanto, a su unión y reacción en el sitio activo de una enzima. A su vez, estos determinarán la afinidad y especificidad de la enzima por diferentes sustratos, y la potencia de los inhibidores enzimáticos (Kirchmair et al., 2012; Basheer et al., 2017).
Aquí, proponemos una nueva enfoque para medir la frecuencia alélica de mutaciones de CYP3A4 en diferentes grupos étnicos. Este enfoque integral tiene el poder de resaltar las mutaciones que prevalecen en grupos étnicos particulares, y combinado con la detección de sustancias químicas que interactúan, p. Ej., Inhibidores de los alimentos, permitirá dilucidar los efectos de mutaciones particulares en la interacción fármaco-alimento, sirviendo como una primera paso hacia la medicina y la nutrición personalizadas. Este trabajo puede crear conciencia sobre la posible importancia clínica de los SNP del CYP3A4 que alteran las proteínas y también sugiere algunas herramientas necesarias para la promoción y aplicación de la medicina de precisión y personalizada.
Materiales y métodos
Análisis de datos y análisis de la base de datos
El conjunto de datos de variantes de CYP3A4 se descargó del navegador gnomAD2 como un archivo CVS. Se utilizó Python 2.7 con paquetes NumPy, pandas y matplotlib para el análisis y la visualización de datos (consulte la hoja de datos complementaria S1). La agrupación jerárquica aglomerativa se realizó utilizando el software Expander 7 (Shamir et al., 2005) con el coeficiente de correlación de rango de Pearson como medida de similitudes y tipo de vinculación completa. Se estableció un umbral de distancia de 0,6 para la agrupación de SNP.
Modelado de polimorfismo in silico
La versión Maestro 2017-2 (Schrodinger, Nueva York, NY, Estados Unidos) se utilizó para la modelado computacional. El modelo de acoplamiento CYP3A4 se construyó como se describió anteriormente (Basheer et al., 2017). En resumen, la estructura cristalina de CYP3A4 (entrada PDB 2V0M) se procesó, modificó y refinó siguiendo los pasos del Asistente de preparación de proteínas. Se generó una cuadrícula de acoplamiento con una restricción de coordinación de metal para el Fe2 + en el grupo hemo basándose en el centroide de ketoconazol en el sitio de unión original en la estructura cristalina. Se seleccionaron siete mutaciones para simulaciones de acoplamiento, una como representante de cada grupo étnico (Tablas 1, 2). Para cada proteína variante, se introdujo una mutación puntual única antes de los pasos de preparación de la proteína. Se generaron estructuras 3D de ligandos basándose en estructuras 2D de PubChem3 y se prepararon para el acoplamiento utilizando la tarea LigPrep.El campo de fuerza OPLS3 y las opciones de deslizamiento predeterminadas para la precisión estándar se aplicaron para el modelo de acoplamiento, con la excepción de que se utilizó la restricción de coordinación de metal, así como 30 poses para el número de poses a incluir y 10 poses para el número de poses para escribir fuera. Para cada ligando, se seleccionó el resultado de acoplamiento con la puntuación más baja de Glide emodel.

Tabla 1. SNP representativos seleccionados para siete grupos étnicos.

Tabla 2. Frecuencia (%) de mutaciones seleccionadas por grupo étnico.
Resultados
La base de datos de agregación del genoma (gnomAD; ver la nota al pie del texto 2) agrega datos de secuenciación del genoma y del exoma de una amplia variedad de proyectos de secuenciación a gran escala. Incluye datos de 125.748 secuencias de exoma y 15.708 secuencias de genoma completo de 141.456 individuos no relacionados que representan siete poblaciones étnicas (Lek et al., 2016). La base de datos GnomAD presenta 856 variantes de CYP3A4, de las cuales 397 son intrónicas y hasta 459 son exónicas. De los SNP exónicos, 312 son mutaciones sin sentido, lo que indica que afectan la estructura de la proteína. El gen CYP3A4 tiene una longitud de 34.205 pb. Sus 13 exones comprenden una región codificante de 1512 pb que produce una proteína de 504 aminoácidos. Los 412 SNP exónicos con posiciones únicas en este gen dan como resultado una densidad de SNP exónicos de 272 / kpb (Tabla complementaria S1).
El cálculo de frecuencias de alelos diferenciales por grupo étnico revela que algunas poblaciones exhiben frecuencias más altas de mutaciones (Figura 1A). La mayoría de las mutaciones CYP3A4 en la población europea son de hecho raras, como se piensa comúnmente, mientras que las mutaciones en otras poblaciones, como África y Asia oriental, son mucho más prevalentes (Tabla complementaria S2).
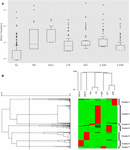
Figura 1. Análisis de SNP de sentido erróneo de CYP3A4 en siete poblaciones distintas. (A) Diagrama de caja a escala logarítmica de frecuencias alélicas. Los recuadros representan el rango intercuartílico (IQR), las líneas azules representan las medianas, los bigotes representan datos dentro de 1,5 IQR y los valores atípicos se muestran como círculos pequeños. (B) Agrupación jerárquica de frecuencias alélicas. Cada fila representa un único SNP. Cada columna representa una población étnica distinta. La frecuencia alélica de los SNP en cada una de las poblaciones está representada por el color de la celda correspondiente en el archivo de matriz. El verde y el rojo representan frecuencias bajas y altas, respectivamente. El dendrograma superior muestra similitudes en el patrón de frecuencia de alelos entre cada grupo de sujetos. El dendrograma de la izquierda representa la agrupación de genes en dos grupos. La línea discontinua representa el umbral de distancia de 0,6 utilizado para dividir en grupos. UE: europeo (no finlandés; n = 64.603), FIN: europeo (finlandés; n = 12.562), ASH J: judío asquenazí (n = 5.185), LTN: latino (n = 17.720), AFR: africano (n = 12,487), E ASN – Este de Asia (n = 9,977), S ASN – Sur de Asia (n = 64,603).
Usamos agrupamiento jerárquico para agrupar variantes con patrones de frecuencia similares. Nuestro análisis de datos arrojó siete grupos distintos (Figura 1B). Además, se observa claramente que los SNP de alta frecuencia en cada grupo son característicos de una población específica. El análisis de agrupamiento jerárquico de los grupos étnicos apoya la asociación entre la varianza genética y la etnia al agrupar etnias relacionadas, como los asiáticos del sur y del este, así como los europeos finlandeses y no finlandeses.
Se utilizó un modelo computacional para evaluar la posible influencia de mutaciones puntuales en CYP3A4 sobre su capacidad para unirse a sustratos e inhibidores. CYP3A4 puede oxidar una amplia gama de compuestos endógenos y xenobióticos. En este caso, se seleccionó el ketoconazol como fármaco representativo y un inhibidor específico muy eficaz; se seleccionaron androstenediona y testosterona como hormona endógena representativa; y demetoxicurcumina y epigalocatequina se seleccionaron como representantes de los bioactivos dietéticos. Se construyó un modelo de acoplamiento para predecir las poses de unión de los compuestos seleccionados en el sitio de unión CYP3A4. El modelo se validó primero restaurando con éxito la pose del ketoconazol en el sitio de unión, con una RMSD de 1,52 Å en relación con la estructura cristalina original. Se diseñaron siete proteínas mutantes basándose en la estructura cristalina de la proteína de tipo salvaje (Figura complementaria S1). Para cada grupo étnico, se seleccionó como representativa la mutación única más frecuente. El efecto de mutaciones individuales sobre la unión del sustrato se evaluó basándose en la comparación entre las poses de acoplamiento sobre la proteína nativa y sobre proteínas variantes. Los cambios en las poses de acoplamiento en términos de RMSD se resumen en la Tabla 3.

Tabla 3. RMSD relativo al WT de ligandos de acoplamiento a sitios de unión de siete variantes de CYP3A4.
Se encontró que el efecto de CYP3A4 SNP sobre la unión del sustrato es específico de mutación-sustrato. Solo en unos pocos casos las mutaciones provocaron un cambio en la posición de unión de un ligando en el bolsillo de unión. La postura de acoplamiento de testosterona fue la misma en las siete variantes probadas. Las variantes E262K, D174H y K168N no provocaron un cambio de postura de unión en ninguna de las moléculas probadas. Sin embargo, las mutaciones L373F y T163A cambiaron la posición de unión de la androstenediona para que se colocara paralela al grupo hemo en lugar de perpendicular a él, como en la proteína WT. Además, la androstenediona se rotó de modo que el grupo ciclopentanona esté ubicado próximo al hemo, en lugar del grupo ciclohexanona en la proteína WT. Las mutaciones S222P y L293P causaron solo una pequeña rotación en la posición de unión de la androstenediona (Figura 2A). De todas las mutaciones examinadas, sólo S222P provocó cambios sustanciales en las posturas de acoplamiento de ketoconazol y demetoxicurcumina en el sitio de unión (Figuras 2B, C); mientras que para la epigalocatequina, la mutación de cambio de pose fue L373F (Figura 2D).
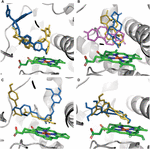
Figura 2. Modelos de ligandos acoplados en el sitio de unión de CYP3A4. (A) ketoconazol, (B) androstenediona, (C) demetoxicurcumina y (D) epigalocatequina. El sitio de unión a proteínas está representado por cintas grises; el hemo está representado por palos verdes, las poses de acoplamiento en la proteína WT y en S222P y L373F los mutantes se muestran como palos naranjas, azules y violetas, respectivamente. Las poses de acoplamiento de androstenediona en las variantes L293P y T136A se superponen con las poses en las variantes S222P y L373F, respectivamente.
Discusión
El citocromo P450 3A4 es la principal enzima responsable de las interacciones entre alimentos y medicamentos. La investigación actual sobre mutaciones en CYP3A4 se ha centrado en unas pocas docenas de SNP encontrados en estudios designados (Sata et al., 2000; Dai et al., 2001; Eiselt et al., 2001; Hsieh et al., 2001; Lamba et al. ., 2002; Murayama et al., 2002). Como se demuestra aquí, representan la punta de un iceberg considerando la prevalencia y los resultados potenciales de las mutaciones CYP3A4. La abundancia de proyectos de secuenciación de grandes genomas y exomas ha abierto una nueva vía para la identificación de muchas mutaciones desconocidas. Aquí, mostramos que las mutaciones presentadas anteriormente son solo la punta del iceberg, al demostrar 856 mutaciones existentes en CYP3A4, de las cuales un tercio modifica la estructura de la proteína. Utilizando una cohorte de 141.456 individuos no emparentados, se calcularon las frecuencias alélicas precisas de mutaciones de CYP3A4 para siete etnias distintas. Hasta donde sabemos, este es el estudio más grande y completo de grandes datos sobre mutaciones exónicas del CYP3A4 y sus frecuencias alélicas en diferentes poblaciones, publicado hasta la fecha.
Las enzimas CYP3A4 polimórficas pueden ser muy importantes para explicar diferencias en la eficacia y toxicidad de los fármacos entre diferentes individuos. Las mutaciones en el gen CYP3A4 pueden conducir a una actividad enzimática anulada, reducida, alterada o aumentada. Las mutaciones exónicas pueden modificar la actividad enzimática, como se ha demostrado en algunos estudios clínicos con sustratos seleccionados. Algunos casos de metabolismo alterado debido a SNP en CYP3A4 ya se han descrito en la literatura (Eiselt et al., 2001; Miyazaki et al., 2008). A pesar de la importancia funcional y la relevancia clínica de los SNP en CYP3A4 y posiblemente debido a su frecuencia relativamente baja identificada en la población general, el polimorfismo en CYP3A4 no ha recibido la atención que merece.
Aquí, siete mutaciones sirvieron para predecir el efecto de los SNP sobre la orientación de unión a sustrato e inhibidor. En la literatura, el polimorfismo CYP3A4 divide a la población general en tres grupos: metabolizadores lentos, metabolizadores normales y metabolizadores rápidos, según los SNP intrónicos que modifican los niveles de expresión en lugar de la estructura (Zanger y Schwab, 2013). Nuestros cálculos sugieren una clasificación adicional: los metabolizadores alterados. Algunas mutaciones propuestas por nuestro modelo virtual provocarían un cambio en la orientación de unión de los ligandos individuales. Se esperaría que estos cambios disminuyan la probabilidad de oxidación enzimática debido a una mayor distancia del hemo, o que conduzcan a productos que de otro modo no serían evidentes durante las pruebas de toxicidad realizadas como parte del proceso de desarrollo de fármacos. Sin embargo, como predice nuestro modelo, para la mayoría de los sustratos las mutaciones del CYP3A4 son benignas.
La posición modificada de un sustrato en el bolsillo de unión debido al cambio estructural de la proteína es solo un posible mecanismo por el cual una mutación podría cambiar la actividad de una proteína. . El anclaje deficiente de la proteína a la membrana, los canales conductores del sustrato dañados y la salida comprometida de los productos presentan mecanismos adicionales para un cambio mutacional en la actividad de una proteína. Como se muestra aquí, el efecto de cada mutación es específico del sustrato.Determinar qué combinaciones de sustratos y mutaciones pueden modificar la actividad enzimática, utilizando métodos tradicionales in vitro es laborioso, enfatizando la necesidad de herramientas virtuales predictivas para resolver este complejo rompecabezas.
Interés público y profesional por la medicina personal y de precisión está creciendo rápidamente. La predicción del metabolismo farmacológico modificado basada en el polimorfismo individual en CYP3A4 parece ser solo una cuestión de tiempo. Aquí, proponemos que los distintos grupos étnicos tengan conjuntos únicos de SNP CYP3A4. De hecho, la etnia puede servir como un primer paso factible en la medicina personalizada, antes de la implementación de una prueba de ADN individual para todos. Curiosamente, el origen étnico tiene una implicación más para el metabolismo del fármaco CYP3A4, ya que es un factor importante en la determinación de las elecciones alimentarias y los hábitos alimentarios. Se puede sugerir que los regímenes terapéuticos deben diseñarse específicamente para cada grupo étnico, al menos para los fármacos que son altamente metabolizados por CYP3A4. Esto destaca las oportunidades para aprovechar e integrar bases de datos y aprendizaje profundo para identificar cómo los SNP, la etnia, los compuestos dietéticos y los medicamentos modifican la actividad de CYP3A4 y el éxito de un régimen médico.
Disponibilidad de datos
En este estudio se analizaron conjuntos de datos disponibles públicamente. Estos datos se pueden encontrar aquí: http://gnomad.broadinstitute.org/gene/ENSG00000160868.
Contribuciones de los autores
Todos los autores enumerados han realizado una contribución sustancial, directa e intelectual a la trabajo y lo aprobó para su publicación.
Declaración de conflicto de intereses
Los autores declaran que la investigación se llevó a cabo en ausencia de relaciones comerciales o financieras que pudieran interpretarse como una conflicto de intereses.
Material complementario
FIGURA S1 | Modelo de cinta 3D de CYP3A4 y la ubicación de los aminoácidos mutados en las siete proteínas variantes diseñadas para el acoplamiento. El hemo se representa como barras verdes, el Fe2 + se representa como una esfera roja, los SNP utilizados en el análisis in silico se representan como áreas rojas en la cinta y los grupos R de aminoácidos mutados en modelos variantes se muestran explícitamente como barras gris claro.
TABLA S1 | Tipos de SNP CYP3A4 en una población de 141, 456 individuos no relacionados que representan 7 poblaciones étnicas.
TABLA S2 | CYP3A4 SNP por grupo étnico.
Notas al pie
- ^ www.pharmvar.org
- ^ https://gnomad.broadinstitute.org
- ^ https://pubchem.ncbi.nlm.nih.gov