Az Angelman-szindróma (AS) egy ritka neurogenetikai rendellenesség, amely körülbelül 15 000 embert érint – világszerte körülbelül 500 000 embert. Az AS-ben szenvedő gyermekek és felnőttek általában egyensúlyi problémákkal, motoros károsodással és gyengítő rohamokkal küzdenek. Néhány egyén soha nem jár. A legtöbb nem beszél. A megszakított alvási ciklusok szintén komoly kihívást jelenthetnek az egyén és a gondozó (k) számára. Az AS-ben szenvedő egyének folyamatos ellátást igényelnek, és nem képesek önálló életet élni. Normális várható élettartamuk van. Ez az élet ma az Angelman-szindrómában szenvedők számára, de a remény itt van. A tudósok úgy vélik, hogy az AS-nek a legnagyobb a gyógyulási lehetősége, összehasonlítva más neurogenetikai rendellenességekkel, és a FAST-nak (Alapítvány az Angelman-szindróma terápiájához) már készen van a terv ennek megvalósítására.
Típusok okai tesztek & Diagnosztikai erőforrások
Angelman-szindróma típusai
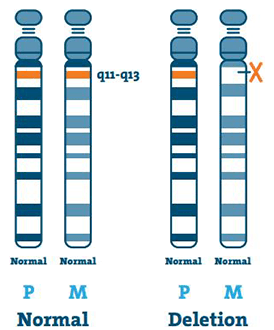
Törlés ( 65-75%)
A DNS (dezoxiribonukleinsav) a kromoszómák fő összetevője. Egyedi genetikai kódunkat tartalmazza. Az AS-ben szenvedő legtöbb embernek hiányzik egy DNS-része az 15q11-13 régióból az anyai 15. kromoszómán.
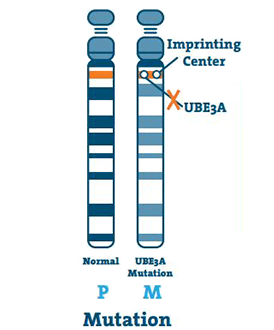
Mutáció (5 -11%)
Ez akkor fordul elő, ha az UBE3A gén DNS-ben van egy kis rendellenesség. A mutáció a gén bárhol előfordulhat.
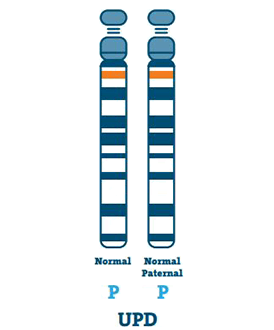
Egyszülői diszómia (3-7%)
Az UPD-vel rendelkező egyénnek az apja és az anyja helyett egy-egy kópia van a 15. kromoszómából. Az UPD általában akkor történik, ha a petesejtben nincs 15. kromoszóma.
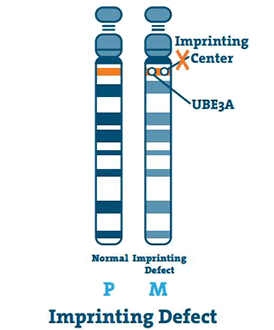
Imprinting Defect (< 3%)
Az imprinting központ egy kis DNS szakasz, amely a kromoszóma q11-13 régiójában helyezkedik el. Ritka esetekben az anya 15. kromoszómája üres, és a központ lemásolja az apa 15. kromoszómáját. Ez egy lenyomathiba.
Jellemzők
Az AS tipikus jellemzői általában nem nyilvánulnak meg születés. A rendellenességben szenvedők csecsemőként táplálkozási nehézségekkel küzdenek, és észrevehetően késik a fejlődés 6-12 hónapos koruk körül. Intenzív terápiákra van szükségük a funkcionális készségek fejlesztéséhez. Az AS minden versenyt és mindkét nemet érint. Gyakran tévesen diagnosztizálják autizmusnak vagy agyi bénulásnak.
Viselkedési tulajdonságok
Az Angelman-szindrómában szenvedőknek van néhány különös viselkedési tulajdonságuk, köztük a boldog viselkedés, amelyet gyakori nevetés, mosolygás és izgatottság jellemez. Sok AS-ban szenvedő ember el van ragadtatva a víz iránt, és nagy örömet szerez az olyan tevékenységek iránt, mint az úszás és a fürdés. működését az UBE3A génben az anya 15. kromoszómáján. Az embereknek két kromoszóma-csoportjuk van – egyet az anyától, egyet pedig az apától örököltek. Egy tipikus embernél az anyai örökölt UBE3A gén aktív, míg az apától örökölt gén kópiája elnémul az agyunk idegsejtjeiben – ezt a jelenséget nevezik imprintingnek. AS-val rendelkező embereknél ez az anyai gén nem végzi a dolgát, és ez hatással van a Messenger RNS-re (mRNS).
![]()
Mi az mRNS?
Az mRNS a test FedEx-je. DNS -ünk az mRNS-t használja szállítási szolgáltatásként, hogy tervrajzokat küldjön sejtjeink fehérje-összeszerelő gyáraiba. Az AS-ben szenvedő emberek mutációval, delécióval vagy egyéb hibával rendelkeznek az UBE3A génjükben, ami megszakítja ezt a szállítási szolgáltatást. Ennek eredményeként neuronjaik nem termelnek funkcionális UBE3A fehérjét, és ez váltja ki az AS tüneteit. Ez a fehérje az, ami segít járni, beszélgetni és más mindennapi feladatokat végrehajtani.
![]()
Genetikai?
A legtöbb esetben az Angelman-szindróma nem öröklődik – különösen azok, amelyeket deléció vagy UPD okoz. Ehelyett ezek a genetikai változások véletlenszerű eseményként fordulnak elő a reproduktív sejtek kialakulása vagy az embrionális fejlődés korai szakaszában.
Tesztek & Diagnózis
Ismerje meg azokról a kritériumokról, amelyeket az orvosok alkalmaznak annak megállapítására, hogy egy személynek van-e Angelman-szindróma, és olvassa el az elérhető teszteket és a gyakori téves diagnózisokat. Tesztelés
![]()
Diagnózis

Csatlakozzon közösségünkhöz
Kapcsolatba léphet más emberekkel, akiknek gyermeke vagy szeretettje van Angelman-szindrómában.
