In questa pagina:
- Cos’è l’acromegalia?
- Quanto è comune l’acromegalia?
- Chi ha maggiori probabilità di sviluppare acromegalia?
- Quali sono le complicazioni dell’acromegalia?
- Quali sono i sintomi dell’acromegalia?
- Quali sono le cause dell’acromegalia?
- In che modo i medici diagnosticano l’acromegalia?
- In che modo i medici trattano l’acromegalia?
- Studi clinici per l’acromegalia
Che cos’è l’acromegalia?
L’acromegalia è un disturbo che si verifica quando il tuo corpo produce una quantità eccessiva di ormone della crescita (GH). Prodotto principalmente nella ghiandola pituitaria, il GH controlla la crescita fisica del corpo. Negli adulti, una quantità eccessiva di questo ormone fa aumentare le dimensioni delle ossa, della cartilagine, degli organi del corpo e di altri tessuti. Cambiamenti comuni nell’aspetto includono naso, orecchie, mani e piedi ingrossati o gonfi.
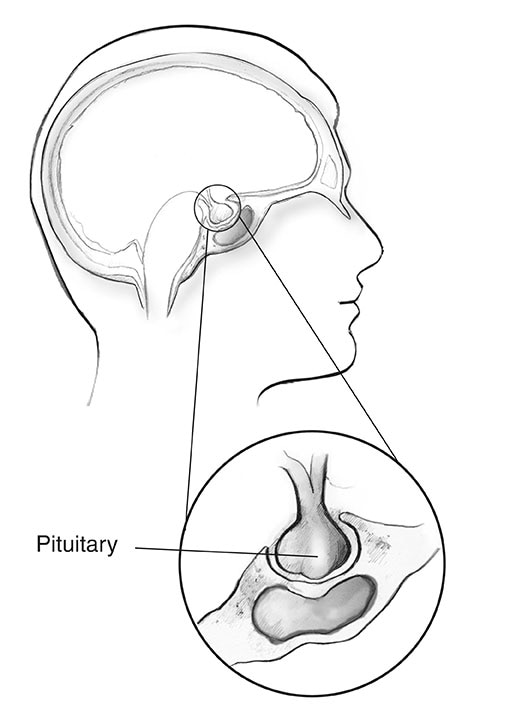
Quanto è comune l’acromegalia?
L’acromegalia è rara. Gli scienziati stimano che da 3 a 14 persone su 100.000 sia stata diagnosticata acromegalia.1
Chi ha maggiori probabilità di sviluppare acromegalia?
L’acromegalia viene diagnosticata più spesso nelle persone di mezza età adulti, ma i sintomi possono comparire a qualsiasi età. Nei bambini, una quantità eccessiva di ormone della crescita causa una condizione chiamata gigantismo piuttosto che acromegalia. Il gigantismo si verifica quando l’eccesso di GH inizia prima della fine della pubertà, quando le piastre di crescita dei bambini si fondono o si chiudono. Avere troppo GH prima che le piastre di crescita si chiudano fa sì che i bambini crescano in altezza.
Quali sono le complicanze dell’acromegalia?
L’acromegalia è curabile nella maggior parte delle persone. Ma poiché i sintomi si manifestano lentamente, possono svilupparsi problemi di salute prima che il disturbo venga diagnosticato e trattato.
I problemi di salute possono includere
- diabete di tipo 2
- pressione alta
- malattie cardiache
- apnea notturna
- artrite
- sindrome del tunnel carpale
- altre condizioni che colpiscono il ossa e muscoli
Le persone con acromegalia hanno anche un aumentato rischio di polipi del colon, che possono svilupparsi in cancro al colon se non vengono rimossi.
Alcune persone con acromegalia possono avere un condizione genetica che può portare a sviluppare tumori in diverse parti del corpo. Un aumento del GH può causare la crescita di questi altri tumori.
L’acromegalia non trattata può portare a seri problemi di salute e morte precoce. Ma se trattati con successo, i sintomi generalmente migliorano e possono scomparire del tutto. L’aspettativa di vita può tornare alla normalità.2
Quali sono i sintomi dell’acromegalia?
I sintomi dell’acromegalia possono variare da persona a persona. Cambiamenti comuni nell’aspetto fisico includono
- mani e piedi che diventano più grandi e gonfi: potresti notare un cambiamento nella misura dell’anello o della scarpa, in particolare la larghezza delle scarpe
- labbra, naso e la lingua diventa più grande
- cambiamenti ossei: la fronte e la mascella inferiore sporgono in fuori, il ponte del naso si allarga e lo spazio tra i denti aumenta
- la pelle diventa spessa, ruvida e grassa
- aumento della sudorazione e dell’odore della pelle
- la voce diventa più profonda
- le etichette della pelle, piccole escrescenze di pelle solitamente color carne che hanno una superficie rialzata, possono diventare più grandi o più scure
Altri sintomi comuni includono
- mal di testa
- dolori articolari
- problemi di vista
Cosa causa l’acromegalia?
L’acromegalia si sviluppa quando la ghiandola pituitaria rilascia una quantità eccessiva di GH nel corpo per un lungo periodo di tempo. Quando il GH entra nel sangue, questo segnala al fegato di produrre un altro ormone, chiamato fattore di crescita simile all’insulina I (IGF-I). IGF-I è l’ormone che effettivamente fa crescere le ossa e il tessuto corporeo. Livelli elevati di questo ormone causano anche cambiamenti nel modo in cui il corpo elabora il glucosio nel sangue (zucchero nel sangue) e i lipidi (grassi), che possono portare a diabete di tipo 2, ipertensione e malattie cardiache.
In più di 9 casi su 10, l’acromegalia è causata da un tumore della ghiandola pituitaria, chiamato adenoma ipofisario.3 Più raramente, la causa potrebbe essere un tumore in un’altra parte del corpo.
Sebbene gli scienziati non Non so cosa causi lo sviluppo di questi tumori, i fattori genetici potrebbero avere un ruolo. Nei giovani adulti, l’acromegalia è stata collegata a difetti in alcuni geni.
I tumori ipofisari
I tumori ipofisari sono quasi sempre benigni o non cancerosi. Alcuni tumori crescono lentamente e i sintomi di un eccesso di GH potrebbero non essere notati per molti anni. Altri tumori possono crescere rapidamente.
A seconda delle sue dimensioni e della sua posizione, il tumore può premere contro altri tessuti ipofisari.I possibili effetti includono
- cambiamenti nelle mestruazioni nelle donne
- disfunzione erettile negli uomini
- cambiamenti nell’ormone tiroideo, che possono influenzare il peso, i livelli di energia, i capelli e la pelle
- diminuisce il cortisolo, che può causare perdita di peso, vertigini, stanchezza, pressione sanguigna bassa e nausea
Anche un tumore di grandi dimensioni può premere contro le parti vicine del cervello. Questo può portare ad altri sintomi, come mal di testa e problemi di vista.
Alcuni tumori ipofisari che creano l’ormone della crescita possono anche aumentare i livelli di altri ormoni nel corpo. Ad esempio, il tumore può produrre prolattina, l’ormone che spinge le ghiandole mammarie a produrre latte. Questo può portare alla secrezione di latte materno nelle donne.
Tumori non ipofisari
Raramente, l’acromegalia è causata da tumori situati nell’ipotalamo, una piccola area del cervello vicino alla ghiandola pituitaria, pancreas , polmoni o altre parti del torace o dell’addome. Alcuni di questi tumori producono essi stessi l’ormone della crescita. Ma più spesso, i tumori producono l’ormone di rilascio dell’ormone della crescita (GHRH), un ormone che segnala alla ghiandola pituitaria di produrre l’ormone della crescita.
Come fanno i medici a diagnosticare l’acromegalia?
Esami del sangue
Il più delle volte i medici diagnosticano l’acromegalia ordinando due esami del sangue che aiutano a determinare se il tuo corpo sta producendo troppo GH.
- Test IGF. I livelli di GH nel sangue possono cambiare durante il giorno. Un modo affidabile per monitorare il GH nel corpo è misurare il livello di IGF-I nel sangue. Nella maggior parte dei casi, un alto livello di IGF-I suggerisce che hai acromegalia.
- Test di tolleranza al glucosio orale. Per confermare la diagnosi, il medico prescriverà un test di tolleranza al glucosio orale. Per questo test, berrai un liquido zuccherino. Un professionista della salute esaminerà quindi il tuo sangue ogni mezz’ora per 2 ore per misurare i livelli dell’ormone della crescita. Lo zucchero nella bevanda normalmente farà diminuire i livelli di GH. Ma se il tuo corpo produce una quantità eccessiva di ormone, questi livelli non scenderanno abbastanza, confermando così la diagnosi di acromegalia.
Esami di imaging
Se il sangue i test confermano che il tuo corpo sta producendo troppo GH, il tuo medico condurrà test di imaging per individuare e misurare il tumore che potrebbe causare il problema. Due test comunemente usati sono
- Risonanza magnetica per immagini. Il test preferito per visualizzare un tumore ipofisario è la risonanza magnetica per immagini (MRI). La risonanza magnetica utilizza onde radio e magneti per creare immagini dettagliate dei tuoi organi interni e dei tessuti molli senza raggi X.
- Tomografia computerizzata. Se una risonanza magnetica non è una buona opzione per te (ad esempio, se hai un pacemaker o un altro impianto con metallo), il tuo medico può ordinare invece una tomografia computerizzata (TC). La scansione TC utilizza una combinazione di raggi X e tecnologia informatica per creare immagini dei tuoi organi e di altre parti interne del tuo corpo.
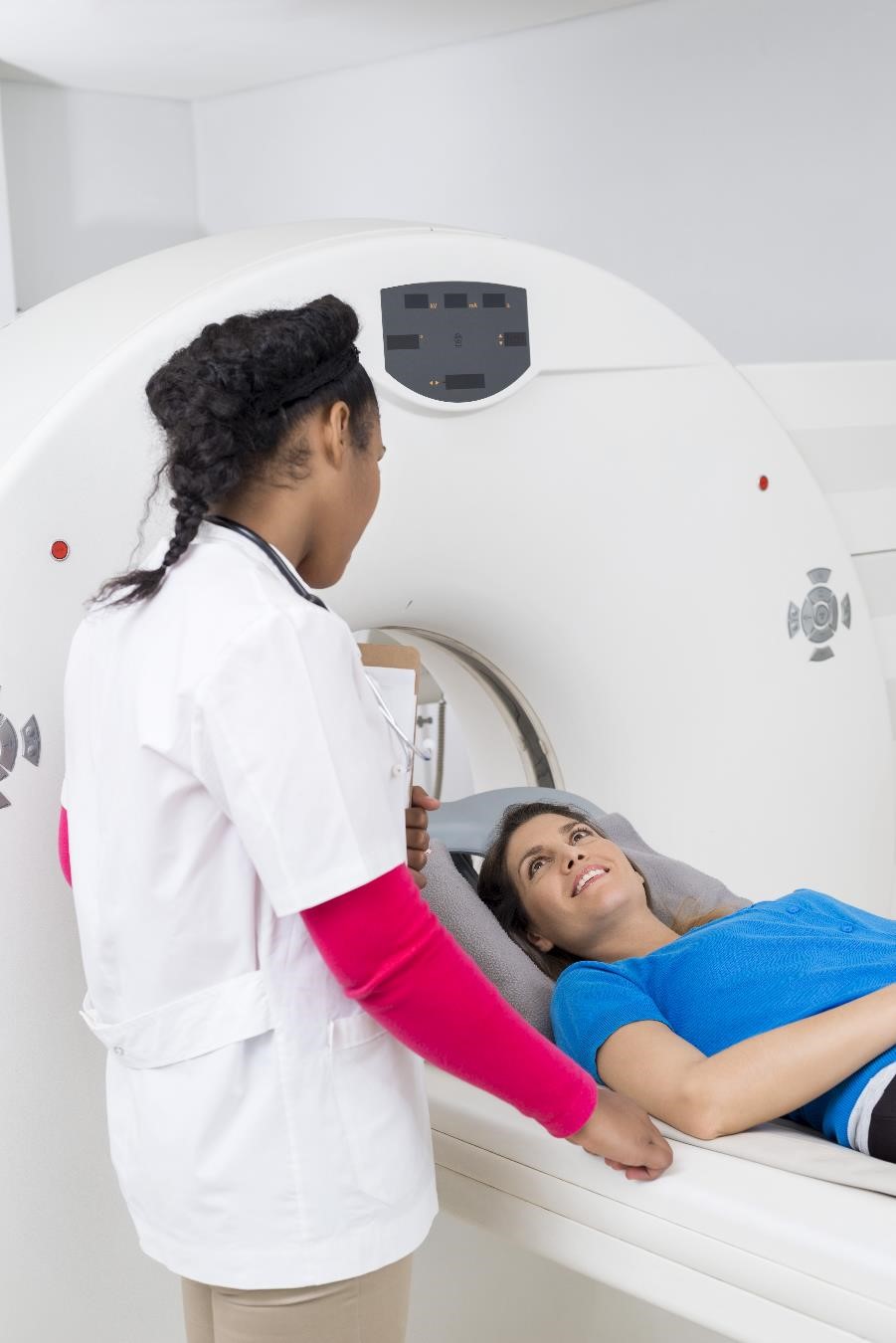
Se il test di imaging non funziona Per trovare un tumore ipofisario, il medico cercherà i tumori non ipofisari come causa dei tuoi livelli elevati di GH.
In che modo i medici trattano l’acromegalia?
Le opzioni di trattamento includono chirurgia, farmaci e radioterapia. Gli obiettivi del trattamento sono controllare le dimensioni del tumore, riportare i livelli di GH e IGF-I alla normalità, migliorare i sintomi e gestire i problemi di salute correlati. Nessun singolo trattamento è giusto per tutti. Il tuo medico ti consiglierà un piano di trattamento adatto a te, a seconda di fattori quali età, dimensioni del tumore, gravità dei sintomi, livelli di GH e IGF-I e stato di salute.
Chirurgia
I medici possono rimuovere la maggior parte dei tumori ipofisari utilizzando un metodo chiamato chirurgia transfenoidale. L’operazione viene eseguita attraverso il naso e il seno sfenoidale, uno spazio cavo nel cranio dietro i passaggi nasali e sotto il cervello. Due approcci a questo intervento sono
- con un microscopio, uno strumento di ingrandimento
- con un endoscopio, un tubo sottile e illuminato con una minuscola fotocamera
In entrambi gli approcci, il chirurgo utilizza l’imaging MRI avanzato per scansionare l’area intorno al tumore prima dell’intervento chirurgico. Poi fa un piccolo taglio all’interno della tua narice per visualizzare l’area e rimuovere il tumore usando piccoli strumenti speciali. Nella chirurgia microscopica, il chirurgo utilizza un microscopio per ingrandire l’area. Nella chirurgia endoscopica, invece, una telecamera per endoscopio invia le immagini a un monitor televisivo. Rischi e risultati sono simili per entrambi gli approcci.3
Quando il tumore che sta creando troppo GH non si trova nella ghiandola pituitaria, vengono utilizzati altri tipi di intervento chirurgico per rimuovere il tumore. La rimozione di questi tumori non ipofisari abbassa anche i livelli di GH e migliora i sintomi dell’acromegalia.
Rischi. Le complicanze della chirurgia possono includere sanguinamento, perdite di liquido cerebrospinale, meningite, squilibrio di sodio (sale) e acqua e bassi livelli di ormoni ipofisari.3
Risultati.L’intervento è considerato un successo se i livelli ematici di GH e IGF-I tornano alla normalità dopo 12 settimane. Il tasso di guarigione subito dopo l’intervento chirurgico è di circa l’85% per i tumori piccoli e dal 40 al 50% per i tumori di grandi dimensioni.3
Quando ha successo, l’intervento chirurgico allevia la pressione sulle aree vicine del cervello e fa scendere i livelli di GH a destra lontano. Il gonfiore dei tessuti molli può migliorare entro pochi giorni, ma i cambiamenti facciali possono richiedere più tempo per migliorare.
La chirurgia ha più successo nelle persone con tumori ipofisari più piccoli. Il successo dipende in gran parte dall’abilità e dall’esperienza del chirurgo, nonché dalla posizione del tumore. Anche chirurghi esperti potrebbero non essere in grado di rimuovere il tumore se è troppo vicino a parti del cervello in cui l’intervento chirurgico sarebbe rischioso. Tuttavia, i chirurghi potrebbero essere in grado di rimuovere parte del tumore.
Trattamenti postchirurgici. Nella maggior parte dei casi, i livelli di GH e IGF-I migliorano ma non tornano alla normalità. Se i livelli di questi ormoni sono ancora troppo alti o iniziano a salire di nuovo, potrebbe essere necessario un ulteriore trattamento. Molto spesso, ciò comporterà l’assunzione di medicinali. In alcuni casi, il medico può raccomandare un secondo intervento chirurgico.
Medicinali
Attualmente, tre tipi di medicinali vengono utilizzati per trattare l’acromegalia, ma non sono una cura. I medicinali possono essere usati da soli o in combinazione tra loro.
Analoghi della somatostatina. I medicinali più spesso usati per trattare l’acromegalia sono chiamati analoghi della somatostatina (SSA). Questi farmaci frenano il rilascio di GH e possono anche ridurre le dimensioni del tumore ipofisario. Diversi studi hanno dimostrato che questi farmaci sono sicuri ed efficaci per il trattamento a lungo termine. I farmaci vengono somministrati per iniezione, ma gli scienziati stanno attualmente studiando altre opzioni, come le pillole.4 Gli effetti collaterali più comuni degli SSA sono crampi, gas e diarrea. Questi effetti sono generalmente lievi e scompaiono nel tempo. Alcune persone possono sviluppare calcoli biliari che di solito non causano sintomi. La caduta dei capelli è possibile e, in rari casi, permanente. Il controllo della glicemia di solito migliora ma, raramente, può peggiorare.
Agonisti della dopamina. Questi farmaci inibiscono la produzione di GH e la crescita del tumore, ma non così bene come gli SSA. Gli agonisti della dopamina hanno maggiori probabilità di funzionare nelle persone che hanno un lieve eccesso di GH e in quelli che hanno sia acromegalia che iperprolattinemia (troppo dell’ormone prolattina). I medicinali vengono presi per bocca. Gli effetti collaterali possono includere nausea, naso chiuso, stanchezza, mal di testa, vertigini in piedi, incubi e cambiamenti di umore.
Antagonisti dei recettori dell’ormone della crescita. A differenza degli altri due medicinali, gli antagonisti del recettore GH non impediscono al corpo di produrre troppo GH. Invece, impediscono al GH di segnalare al corpo di produrre più IGF-I. Il farmaco viene assunto sotto forma di un’iniezione giornaliera sotto la pelle che i pazienti possono somministrare da soli. Gli effetti collaterali possono includere problemi al fegato.
Radioterapia
La terza opzione di trattamento è la radioterapia, che utilizza raggi X ad alta energia o onde di particelle per uccidere le cellule tumorali. Questo tipo di trattamento può essere raccomandato se la chirurgia non è possibile o non riesce a rimuovere tutto il tessuto tumorale e i farmaci non sono un’opzione o funzionano per te.
Stereotassia. Il tipo preferito di radioterapia è la radioterapia stereotassica, che utilizza l’imaging 3-D per puntare con precisione alte dosi di radiazioni al tumore da varie angolazioni.3 Il trattamento a volte può essere eseguito in una singola sessione, riducendo il rischio di danni alle aree circostanti tessuto. Tuttavia, una singola dose potrebbe non funzionare per tumori molto grandi e tumori situati vicino ai nervi che influenzano la vista.
Convenzionale. La seconda opzione è la radioterapia convenzionale, che prende di mira anche il tumore con fasci esterni. Questo tipo di radioterapia eroga piccole dosi di radiazioni in una serie di trattamenti da 4 a 6 settimane.
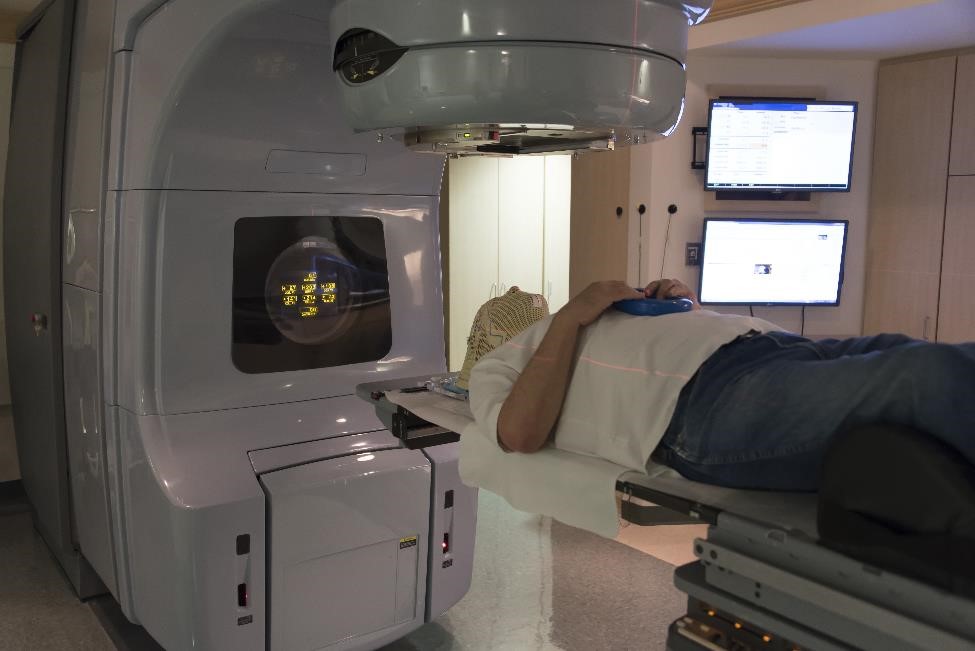
Poiché la radioterapia riduce i livelli di GH e IGF-I nel tempo, potrebbero essere necessari anni prima che questo trattamento migliori sensibilmente i sintomi dell’acromegalia. È probabile che il tuo medico prescriva farmaci mentre aspetti che i livelli di GH e IGF-I tornino alla normalità e che i sintomi migliorino.
Tutte le forme di radioterapia fanno sì che altri ormoni ipofisari diminuiscano lentamente nel tempo. Circa la metà delle persone trattate con radioterapia avrà bisogno di una sostituzione ormonale al termine del trattamento. Le radiazioni possono anche compromettere la fertilità del paziente.
La perdita della vista e le lesioni cerebrali sono complicazioni rare. Raramente, altri tipi di tumori possono svilupparsi molti anni dopo in aree che si trovavano nel percorso del fascio di radiazioni.
Sperimentazioni cliniche per l’acromegalia
Il NIDDK conduce e supporta studi clinici in molti malattie e condizioni, comprese le malattie endocrine. Gli studi cercano di trovare nuovi modi per prevenire, rilevare o curare le malattie e migliorare la qualità della vita.
Cosa sono gli studi clinici per l’acromegalia?
Gli studi clinici e altri tipi di studi clinici fanno parte della ricerca medica e coinvolgono persone come te. Quando ti offri volontario per prendere parte a uno studio clinico, aiuti medici e ricercatori a saperne di più sulla malattia e migliorare l’assistenza sanitaria per le persone in futuro.
I ricercatori stanno studiando molti aspetti dell’acromegalia e del gigantismo, come
- uso di farmaci per trattare il gigantismo nei bambini e negli adolescenti
- fattori genetici che possono causare lo sviluppo di tumori ipofisari e come trattare i tumori e le complicazioni correlate nei bambini e negli adulti
Scopri se gli studi clinici sono adatti a te.
Quali studi clinici sull’acromegalia stanno cercando i partecipanti?
Puoi visualizzare un elenco filtrato di studi clinici sull’acromegalia che sono aperti e di reclutamento su
www.ClinicalTrials.gov. È possibile espandere o restringere l’elenco per includere studi clinici di industria, università e individui; tuttavia, il NIH non esamina questi studi e non può garantire che siano sicuri. Parla sempre con il tuo medico prima di partecipare a uno studio clinico.