Introduzione
Per decenni, è noto che le interazioni cibo-farmaco (FDI) e erbe-farmaci limitano il successo dei trattamenti medici. L’enorme numero di possibili interazioni tra variazioni genetiche, regimi medici e numerosi composti bioattivi presenti negli alimenti e nelle erbe si traduce in una complessità travolgente. Strumenti moderni come l’analisi dei big data, l’apprendimento automatico e la simulazione delle interazioni proteina-ligando possono aiutarci a rispondere a tutta una serie di domande: le scelte alimentari potrebbero contribuire al fallimento dei regimi terapeutici e, se sì, come? Quali alimenti dovrebbero essere consumati prima di assumere un farmaco prescritto? E probabilmente la domanda più interessante: come possiamo utilizzare questi strumenti per prevedere gli IDE personali? Chiaramente, molte risposte risiedono nel metabolismo di farmaci, alimenti ed erbe da parte del citocromo P450 3A4 (CYP3A4) nel fegato e nel tratto digerente (Galetin et al., 2010; Basheer e Kerem, 2015).
La maggior parte dei geni che codificano per gli enzimi CYP sono polimorfici. Ad oggi, la fonte più completa di informazioni dettagliate sugli alleli del CYP è il Pharmacogene Variation Consortium1, in cui sono rappresentati meno di 100 alleli del CYP3A4. Di questi, meno di 40 sono SNP esonici (polimorfismi a singolo nucleotide) che risultano in una sequenza proteica modificata. L’esiguo numero di soggetti in tutti i lavori precedentemente pubblicati sulle mutazioni del CYP3A4 ci fornisce dati limitati sulle frequenze reali delle mutazioni del CYP3A4 nell’intera popolazione e in gruppi definiti.
Non solo le informazioni affidabili sull’incidenza degli SNP sono incomplete , anche le loro implicazioni cliniche sono ancora poco chiare nella maggior parte dei casi (Zanger et al., 2014). Capire quali e quando gli SNP potrebbero avere un significato clinico è un compito estremamente complesso. I test in vitro richiedono molto tempo, sono costosi e praticamente di scarsa rilevanza considerando la grande quantità di mutazioni e il numero infinito di combinazioni cibo-farmaco. I metodi di modellazione molecolare, inclusi i calcoli di docking e free-energy binding, possono servire per prevedere i potenziali effetti degli SNP e di molti composti sul metabolismo mediato dal CYP3A4 (Lewis et al., 1998). Ad esempio, le interazioni non covalenti, idrofobiche, elettrostatiche e di van der Waals contribuiscono tutte all’orientamento di un composto e quindi al suo legame e alla sua reazione nel sito attivo di un enzima. A loro volta, questi determineranno l’affinità e la specificità dell’enzima per diversi substrati e la potenza degli inibitori enzimatici (Kirchmair et al., 2012; Basheer et al., 2017).
Qui, proponiamo un nuovo approccio alla misurazione della frequenza allelica delle mutazioni del CYP3A4 in diversi gruppi etnici. Questo approccio globale ha il potere di evidenziare le mutazioni che sono prevalenti in particolari gruppi etnici e, combinato con lo screening per le sostanze chimiche interagenti, ad esempio, gli inibitori del cibo consentiranno la delucidazione degli effetti di particolari mutazioni sull’interazione farmaco-cibo, fungendo da iniziale passo verso la medicina e la nutrizione personalizzate. Questo lavoro può aumentare la consapevolezza della possibile importanza clinica degli SNP CYP3A4 che alterano le proteine e suggerisce anche alcuni strumenti necessari per la promozione e l’applicazione della medicina di precisione e personalizzata.
Materiali e metodi
Screening del database e analisi dei dati
Il dataset delle varianti CYP3A4 è stato scaricato dal browser2 gnomAD come file CVS. Python 2.7 con NumPy, panda e pacchetti matplotlib è stato utilizzato per l’analisi e la visualizzazione dei dati (vedere il foglio dati supplementare S1). Il clustering gerarchico agglomerativo è stato eseguito utilizzando il software Expander 7 (Shamir et al., 2005) con il coefficiente di correlazione dei ranghi di Pearson come misura delle somiglianze e del tipo di collegamento completo. È stata fissata una soglia di distanza di 0,6 per il raggruppamento di SNP.
In silico Polymorphism Modeling
La versione Maestro 2017-2 (Schrodinger, New York, NY, Stati Uniti) è stata utilizzata per modellazione computazionale. Il modello di docking CYP3A4 è stato costruito come descritto in precedenza (Basheer et al., 2017). In breve, la struttura cristallina del CYP3A4 (file PDB 2V0M) è stata elaborata, modificata e raffinata seguendo i passaggi della procedura guidata di preparazione delle proteine. Una griglia di ancoraggio con un vincolo di coordinazione del metallo per Fe2 + nel gruppo eme è stata generata in base al centroide del ketoconazolo nel sito di legame originale nella struttura cristallina. Sono state selezionate sette mutazioni per le simulazioni di docking, una come rappresentante per ogni gruppo etnico (Tabelle 1, 2). Per ciascuna variante della proteina, è stata introdotta una singola mutazione puntiforme prima delle fasi di preparazione della proteina. Strutture 3D di ligandi sono state generate sulla base di strutture 2D da PubChem3 e preparate per l’attracco utilizzando l’attività LigPrep.Per il modello di attracco sono state applicate il campo di forza OPLS3 e le opzioni Glide predefinite per la precisione standard, con l’eccezione che è stato utilizzato il vincolo di coordinazione del metallo, oltre a 30 pose per il numero di pose da includere e 10 pose per il numero di pose da scrivere su. Per ogni ligando, è stato selezionato il risultato di attracco con il punteggio di Glide emodel più basso.

Tabella 1. SNP rappresentativi selezionati per sette gruppi etnici.

Tabella 2. Frequenza (%) delle mutazioni selezionate per gruppo etnico.
Risultati
Il Genome Aggregation Database (gnomAD; vedere la nota di testo 2) aggrega i dati di sequenziamento dell’esoma e del genoma da un’ampia varietà di progetti di sequenziamento su larga scala. Include dati da 125.748 sequenze di esomi e 15.708 sequenze dell’intero genoma da 141.456 individui non imparentati che rappresentano sette popolazioni etniche (Lek et al., 2016). Il database di GnomAD presenta 856 varianti di CYP3A4, di cui 397 introniche e ben 459 esoniche. Degli SNP esonici, 312 sono mutazioni missenso, indicando che influenzano la struttura della proteina. Il gene CYP3A4 è lungo 34.205 bp. I suoi 13 esoni comprendono una regione codificante di 1.512 bp che produce una proteina di 504 aminoacidi. I 412 SNP esonici con posizioni uniche in questo gene risultano in una densità SNP esonica di 272 / kbp (Tabella supplementare S1).
Il calcolo delle frequenze alleliche differenziali per gruppo etnico rivela che alcune popolazioni mostrano frequenze di mutazioni più elevate (Figura 1A). La maggior parte delle mutazioni del CYP3A4 nella popolazione europea sono infatti rare, come comunemente si pensa, mentre le mutazioni in altre popolazioni, come quella africana e dell’Asia orientale, sono molto più prevalenti (Tabella supplementare S2).
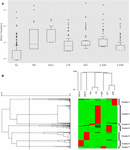
Figura 1. Analisi degli SNP missenso CYP3A4 in sette popolazioni distinte. (A) Box plot in scala logaritmica delle frequenze alleliche. Le caselle rappresentano l’intervallo interquartile (IQR), le linee blu rappresentano le mediane, i baffi rappresentano i dati entro 1,5 IQR e i valori anomali sono mostrati come piccoli cerchi. (B) clustering gerarchico delle frequenze alleliche. Ogni riga rappresenta un singolo SNP. Ogni colonna rappresenta una popolazione etnica distinta. La frequenza allelica degli SNP in ciascuna delle popolazioni è rappresentata dal colore della cella corrispondente nel file della matrice. Il verde e il rosso rappresentano rispettivamente la bassa e l’alta frequenza. Il dendrogramma superiore mostra somiglianze nel modello di frequenza allelica tra ciascun gruppo di soggetti. Il dendrogramma di sinistra rappresenta il raggruppamento dei geni in due gruppi. La linea tratteggiata rappresenta la soglia di 0,6 distanza utilizzata per la suddivisione in gruppi. UE – Europea (non finlandese; n = 64.603), FIN – Europea (Finlandese; n = 12.562), ASH J – Ashkenazi Jewish (n = 5.185), LTN – Latino (n = 17.720), AFR – Africana (n = 12.487), E ASN – Asia orientale (n = 9.977), S ASN – Asia meridionale (n = 64.603).
Abbiamo utilizzato il clustering gerarchico per raggruppare varianti con modelli di frequenza simili. La nostra analisi dei dati ha prodotto sette cluster distinti (Figura 1B). Inoltre, si osserva chiaramente che gli SNP ad alta frequenza in ciascun cluster sono caratteristici di una popolazione specifica. L’analisi gerarchica dei gruppi etnici supporta l’associazione tra varianza genetica ed etnia raggruppando insieme etnie correlate come gli asiatici del sud e dell’est, nonché gli europei finlandesi e non finlandesi.
Per la valutazione è stato utilizzato un modello computazionale la possibile influenza delle mutazioni puntiformi nel CYP3A4 sulla sua capacità di legare substrati e inibitori. Il CYP3A4 è in grado di ossidare un’ampia gamma di composti endogeni e xenobiotici. Qui, il ketoconazolo è stato selezionato come farmaco rappresentativo e un inibitore specifico molto efficiente; androstenedione e testosterone sono stati selezionati come ormone endogeno rappresentativo; e demetossicurcumina ed epigallocatechina sono stati selezionati come rappresentanti di bioattivi dietetici. È stato costruito un modello di docking per prevedere le pose di legame dei composti selezionati nel sito di legame del CYP3A4. Il modello è stato convalidato per la prima volta ripristinando con successo la posa del ketoconazolo nel sito di legame, con un RMSD di 1,52 Å rispetto alla struttura cristallina originale. Sette proteine mutanti sono state progettate in base alla struttura cristallina della proteina wild-type (Figura supplementare S1). Per ogni gruppo etnico, la mutazione unica più frequente è stata selezionata come rappresentativa. L’effetto di singole mutazioni sul legame al substrato è stato valutato sulla base del confronto tra le pose di attracco sulla proteina nativa e sulle proteine varianti. I cambiamenti nelle pose di attracco in termini di RMSD sono riassunti nella Tabella 3.

Tabella 3. RMSD relativo al WT dei ligandi di attracco ai siti di legame di sette varianti di CYP3A4.
L’effetto del CYP3A4 SNP sul legame del substrato è stato trovato essere specifico del substrato della mutazione. Solo in pochi casi le mutazioni hanno causato un cambiamento nella posizione di legame di un ligando nella tasca di legame. La posizione di attracco del testosterone era la stessa in tutte e sette le varianti testate. Le varianti E262K, D174H e K168N non hanno causato un cambiamento della posa di legame in nessuna delle molecole testate. Tuttavia, le mutazioni L373F e T163A hanno modificato la posizione di legame dell’androstenedione in modo che fosse posizionato parallelamente al gruppo eme piuttosto che perpendicolare ad esso, come nella proteina WT. Inoltre, androstenedione è stato ruotato in modo tale che il gruppo ciclopentanone si trovi prossimale all’eme, invece del gruppo cicloesanone nella proteina WT. Le mutazioni S222P e L293P hanno causato solo una piccola rotazione nella posa di legame dell’androstenedione (Figura 2A). Di tutte le mutazioni esaminate, solo S222P ha causato cambiamenti sostanziali nelle posizioni di attracco di ketoconazolo e demetossicurcumina nel sito di legame (Figure 2B, C); mentre per l’epigallocatechina, la mutazione che cambia la posizione era L373F (Figura 2D).
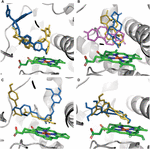
Figura 2. Modelli di ligandi ancorati al sito di legame del CYP3A4. (A) ketoconazolo, (B) androstenedione, (C) demetossicurcumina e (D) epigallocatechina. Il sito di legame delle proteine è rappresentato da nastri grigi; L’eme è rappresentato da bastoncini verdi, le pose di aggancio nella proteina WT e nei mutanti S222P e L373F sono mostrate rispettivamente come bastoncini arancioni, blu e viola. Le pose di ancoraggio androstenedione nelle varianti L293P e T136A si sovrappongono alle pose nelle varianti S222P e L373F, rispettivamente.
Discussione
Il citocromo P450 3A4 è il principale enzima responsabile delle interazioni cibo-farmaco. La ricerca attuale sulle mutazioni nel CYP3A4 si è concentrata su poche dozzine di SNP trovati in studi designati (Sata et al., 2000; Dai et al., 2001; Eiselt et al., 2001; Hsieh et al., 2001; Lamba et al. ., 2002; Murayama et al., 2002). Come dimostrato qui, rappresentano la punta di un iceberg considerando la prevalenza e i potenziali esiti delle mutazioni del CYP3A4. L’abbondanza di progetti di sequenziamento di grandi genomi ed esomi ha aperto una nuova strada per l’identificazione di molte mutazioni sconosciute. Qui, mostriamo che le mutazioni presentate in precedenza sono solo la punta dell’iceberg, dimostrando 856 mutazioni esistenti nel CYP3A4, di cui un terzo modificano la struttura della proteina. Utilizzando una coorte di 141.456 individui non imparentati, sono state calcolate accurate frequenze alleliche delle mutazioni del CYP3A4 per sette etnie separate. Per quanto ne sappiamo, questo è il più ampio e completo studio su dati di grandi dimensioni delle mutazioni esoniche del CYP3A4 e delle loro frequenze alleliche in diverse popolazioni, pubblicato fino ad oggi.
Gli enzimi polimorfici del CYP3A4 possono essere molto importanti per la spiegazione differenze nell’efficacia e nella tossicità del farmaco tra i diversi individui. Le mutazioni nel gene CYP3A4 potrebbero portare ad attività enzimatica abolita, ridotta, alterata o aumentata. Le mutazioni esoniche possono modificare l’attività enzimatica, come è stato dimostrato in alcuni studi clinici con substrati selezionati. Alcuni casi di metabolismo alterato dovuto agli SNP nel CYP3A4 sono già stati descritti in letteratura (Eiselt et al., 2001; Miyazaki et al., 2008). Nonostante l’importanza funzionale e la rilevanza clinica degli SNP nel CYP3A4 e forse a causa della loro frequenza identificata relativamente bassa nella popolazione generale, il polimorfismo nel CYP3A4 non ha ricevuto l’attenzione che merita.
Qui, sette mutazioni sono servite per prevedere l’effetto degli SNP sull’orientamento del legame del substrato e dell’inibitore. In letteratura, il polimorfismo del CYP3A4 divide la popolazione generale in tre gruppi: metabolizzatori lenti, metabolizzatori normali e metabolizzatori rapidi, basati su SNP intronici che modificano i livelli di espressione piuttosto che la struttura (Zanger e Schwab, 2013). I nostri calcoli suggeriscono una classificazione aggiuntiva: i metabolizzatori alterati. Alcune mutazioni proposte dal nostro modello virtuale causerebbero un cambiamento nell’orientamento di legame dei singoli ligandi. Ci si aspetterebbe che questi cambiamenti riducano la probabilità di ossidazione enzimatica a causa dell’aumentata distanza dall’eme, o portino a prodotti che altrimenti non sarebbero evidenti durante i test di tossicità effettuati come parte del processo di sviluppo del farmaco. Tuttavia, come prevede il nostro modello, per la maggior parte dei substrati le mutazioni del CYP3A4 sono benigne.
La posizione modificata di un substrato nella tasca di legame a causa del cambiamento strutturale della proteina è solo uno dei possibili meccanismi con cui una mutazione potrebbe cambiare l’attività di una proteina . L’ancoraggio alterato della proteina alla membrana, i canali danneggiati che conducono il substrato e l’uscita compromessa dei prodotti presentano meccanismi aggiuntivi per un cambiamento mutazionale nell’attività di una proteina. Come mostrato qui, l’effetto di ogni mutazione è specifico del substrato.Determinare quali combinazioni di substrati e mutazioni potrebbero modificare l’attività enzimatica, utilizzando metodi tradizionali in vitro è laborioso, sottolineando la necessità di strumenti virtuali predittivi per risolvere questo puzzle complesso.
Interesse pubblico e professionale per la medicina personale e di precisione sta crescendo rapidamente. La previsione del metabolismo del farmaco modificato sulla base del polimorfismo individuale nel CYP3A4 sembra essere solo una questione di tempo. Qui, proponiamo che gruppi etnici distinti portino set unici di SNP CYP3A4. In effetti, l’etnia può servire come primo passo fattibile nella medicina personalizzata, che precede l’implementazione di uno screening del DNA individuale per tutti. È interessante notare che l’etnia ha un’ulteriore implicazione per il metabolismo dei farmaci CYP3A4, essendo un fattore importante nel determinare le scelte alimentari e le abitudini alimentari. Si può suggerire che i regimi terapeutici dovrebbero essere specificamente progettati per ciascun gruppo etnico, almeno per i farmaci che sono altamente metabolizzati dal CYP3A4. Ciò evidenzia le opportunità per sfruttare e integrare database e deep learning per identificare come SNP, etnia, composti dietetici e farmaci modificano l’attività del CYP3A4 e il successo di un regime medico.
Disponibilità dei dati
I set di dati disponibili pubblicamente sono stati analizzati in questo studio. Questi dati possono essere trovati qui: http://gnomad.broadinstitute.org/gene/ENSG00000160868.
Contributi degli autori
Tutti gli autori elencati hanno dato un contributo sostanziale, diretto e intellettuale al e approvato per la pubblicazione.
Conflitto di interessi
Gli autori dichiarano che la ricerca è stata condotta in assenza di rapporti commerciali o finanziari che potrebbero essere interpretati come un potenziale conflitto di interessi.
Materiale supplementare
FIGURA S1 | Modello a nastro 3D del CYP3A4 e posizione degli amminoacidi mutati nelle sette proteine varianti progettate per l’aggancio. Heme è rappresentato come bastoncini verdi, Fe2 + è rappresentato come una sfera rossa, gli SNP utilizzati nell’analisi in silico sono rappresentati come aree rosse sul nastro e i gruppi R di amminoacidi mutati nei modelli varianti sono mostrati esplicitamente come bastoncini grigio chiaro.
TABELLA S1 | CYP3A4 SNP tipi in una popolazione di 141, 456 individui non imparentati che rappresentano 7 popolazioni etniche.
TABELLA S2 | SNP CYP3A4 per gruppo etnico.
Note a piè di pagina
- ^ www.pharmvar.org
- ^ https://gnomad.broadinstitute.org
- ^ https://pubchem.ncbi.nlm.nih.gov