Sur cette page:
- Qu’est-ce que l’acromégalie?
- Quelle est la fréquence de l’acromégalie?
- Qui est plus susceptible de développer une acromégalie?
- Quelles sont les complications de l’acromégalie?
- Quels sont les symptômes de l’acromégalie?
- Quelles sont les causes de l’acromégalie?
- Comment les médecins diagnostiquent-ils l’acromégalie?
- Comment les médecins traitent-ils l’acromégalie?
- Essais cliniques sur l’acromégalie
Qu’est-ce que l’acromégalie?
L’acromégalie est un trouble qui survient lorsque votre corps produit trop d’hormone de croissance (GH). Produite principalement dans l’hypophyse, la GH contrôle la croissance physique du corps. Chez les adultes, une trop grande quantité de cette hormone entraîne une augmentation de la taille des os, du cartilage, des organes corporels et d’autres tissus. Les changements d’apparence courants incluent le nez, les oreilles, les mains et les pieds élargis ou enflés.
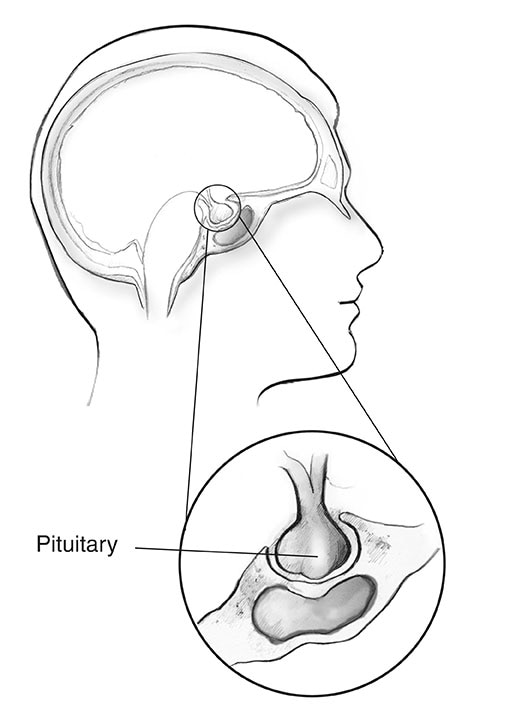
Quelle est la fréquence d’acromégalie?
L’acromégalie est rare. Les scientifiques estiment qu’environ 3 à 14 personnes sur 100 000 ont reçu un diagnostic d’acromégalie.1
Qui est le plus susceptible de développer une acromégalie?
L’acromégalie est le plus souvent diagnostiquée chez les personnes d’âge moyen adultes, mais les symptômes peuvent apparaître à tout âge. Chez les enfants, trop d’hormone de croissance provoque une maladie appelée gigantisme plutôt que acromégalie. Le gigantisme se produit lorsque l’excès de GH commence avant la fin de la puberté, lorsque les plaques de croissance des enfants fusionnent ou se ferment. Avoir trop de GH avant la fermeture des plaques de croissance fait que les enfants grandissent.
Quelles sont les complications de l’acromégalie?
L’acromégalie est traitable chez la plupart des gens. Mais comme les symptômes apparaissent lentement, des problèmes de santé peuvent se développer avant que le trouble ne soit diagnostiqué et traité.
Les problèmes de santé peuvent inclure
- diabète de type 2
- hypertension artérielle
- maladie cardiaque
- apnée du sommeil
- arthrite
- syndrome du canal carpien
- autres affections affectant le les os et les muscles
Les personnes atteintes d’acromégalie ont également un risque accru de polypes du côlon, qui peuvent évoluer en cancer du côlon si elles ne sont pas enlevées.
Certaines personnes atteintes d’acromégalie peuvent avoir un maladie génétique qui peut conduire des tumeurs à se développer dans différentes parties de leur corps. Une augmentation de la GH peut provoquer la croissance de ces autres tumeurs.
Non traitée, l’acromégalie peut entraîner de graves problèmes de santé et la mort prématurée. Mais lorsqu’ils sont traités avec succès, les symptômes s’améliorent généralement et peuvent disparaître complètement. L’espérance de vie peut revenir à la normale.2
Quels sont les symptômes de l’acromégalie?
Les symptômes de l’acromégalie peuvent varier d’une personne à l’autre. Les changements communs dans l’apparence physique incluent
- les mains et les pieds deviennent plus gros et enflés. Vous remarquerez peut-être un changement dans la taille de la bague ou de la chaussure, en particulier la largeur de la chaussure
- les lèvres, le nez et la langue devient plus grande
- changements osseux: le front et la mâchoire inférieure font saillie, l’arête du nez s’agrandit et l’espace entre les dents augmente
- la peau devient épaisse, grossière et grasse
- augmentation de la transpiration et des odeurs cutanées
- la voix devient plus profonde
- les acrochordons (petites excroissances généralement de couleur chair qui ont une surface surélevée) peuvent devenir plus grands ou plus foncés
D’autres symptômes courants incluent
- maux de tête
- douleurs articulaires
- problèmes de vision
Qu’est-ce qui cause l’acromégalie?
L’acromégalie se développe lorsque l’hypophyse libère trop de GH dans le corps pendant une longue période de temps. Lorsque la GH pénètre dans le sang, cela signale au foie de produire une autre hormone, appelée facteur de croissance analogue à l’insuline I (IGF-I). L’IGF-I est l’hormone qui provoque la croissance des os et des tissus corporels. Des niveaux élevés de cette hormone entraînent également des changements dans la façon dont le corps traite la glycémie (sucre dans le sang) et les lipides (graisses), ce qui peut entraîner un diabète de type 2, une pression artérielle élevée et des maladies cardiaques.
En savoir plus plus de 9 cas sur 10, l’acromégalie est causée par une tumeur de l’hypophyse, appelée adénome hypophysaire.3 Plus rarement, la cause peut être une tumeur dans une autre partie du corps.
Bien que les scientifiques ne le soient pas. Je ne sais pas ce qui provoque le développement de ces tumeurs, des facteurs génétiques peuvent jouer un rôle. Chez les jeunes adultes, l’acromégalie a été liée à des anomalies de certains gènes.
Tumeurs hypophysaires
Les tumeurs hypophysaires sont presque toujours bénignes ou non cancéreuses. Certaines tumeurs se développent lentement et les symptômes d’une trop grande quantité de GH peuvent ne pas être remarqués pendant de nombreuses années. D’autres tumeurs peuvent se développer rapidement.
En fonction de sa taille et de son emplacement, la tumeur peut appuyer contre d’autres tissus hypophysaires.Les effets possibles incluent
- des changements dans la menstruation chez la femme
- la dysfonction érectile chez l’homme
- des changements dans l’hormone thyroïdienne, qui peuvent affecter le poids, les niveaux d’énergie, les cheveux et la peau
- diminution du cortisol, ce qui peut entraîner une perte de poids, des étourdissements, de la fatigue, une hypotension artérielle et des nausées
Une tumeur qui grossit peut aussi appuyez contre les parties voisines du cerveau. Cela peut entraîner d’autres symptômes, tels que des maux de tête et des problèmes de vision.
Certaines tumeurs hypophysaires qui créent l’hormone de croissance peuvent également augmenter les niveaux d’autres hormones dans le corps. Par exemple, la tumeur peut produire de la prolactine, l’hormone qui incite les glandes mammaires à produire du lait. Cela peut entraîner un écoulement du lait maternel chez les femmes.
Tumeurs non hypophysaires
L’acromégalie est rarement causée par des tumeurs situées dans l’hypothalamus, une petite zone du cerveau près de l’hypophyse, le pancréas , les poumons ou d’autres parties de la poitrine ou de l’abdomen. Certaines de ces tumeurs fabriquent elles-mêmes l’hormone de croissance. Mais le plus souvent, les tumeurs produisent l’hormone de libération de l’hormone de croissance (GHRH), une hormone qui signale à l’hypophyse de fabriquer de l’hormone de croissance.
Comment les médecins diagnostiquent-ils l’acromégalie?
Analyses sanguines
Les médecins diagnostiquent le plus souvent l’acromégalie en commandant deux tests sanguins qui aident à déterminer si votre corps produit trop de GH.
- Test IGF. Les niveaux de GH dans le sang peuvent changer au cours de la journée. Un moyen fiable de suivre la GH dans le corps consiste à mesurer le niveau d’IGF-I dans le sang. Dans la plupart des cas, un taux élevé d’IGF-I suggère que vous souffrez d’acromégalie.
- Test de tolérance au glucose par voie orale. Pour confirmer le diagnostic, votre médecin vous prescrira un test oral de tolérance au glucose. Pour ce test, vous boirez un liquide sucré. Un professionnel de la santé analysera ensuite votre sang toutes les demi-heures pendant 2 heures pour mesurer les niveaux d’hormone de croissance. Le sucre dans la boisson fera normalement baisser les niveaux de GH. Mais si votre corps produit trop d’hormone, ces niveaux ne diminueront pas suffisamment, confirmant ainsi le diagnostic d’acromégalie.
Tests d’imagerie
Si le sang les tests confirment que votre corps produit trop de GH, votre médecin effectuera des tests d’imagerie pour localiser et mesurer la tumeur qui peut être à l’origine du problème. L’imagerie par résonance magnétique est deux tests couramment utilisés.
- Le test préféré pour visualiser une tumeur hypophysaire est le scan d’imagerie par résonance magnétique (IRM). L’IRM utilise des ondes radio et des aimants pour créer des images détaillées de vos organes internes et tissus mous sans rayons X.
- Tomodensitométrie. Si une IRM n’est pas une bonne option pour vous (par exemple, si vous avez un stimulateur cardiaque ou un autre implant contenant du métal), votre médecin peut demander une tomodensitométrie (TDM) à la place. Le scanner utilise une combinaison de rayons X et de technologie informatique pour créer des images de vos organes et d’autres parties internes de votre corps.
Si le test d’imagerie ne fonctionne pas t trouver une tumeur hypophysaire, votre médecin recherchera des tumeurs non hypophysaires comme cause de vos niveaux élevés de GH.
Comment les médecins traitent-ils l’acromégalie?
Les options de traitement comprennent la chirurgie, les médicaments et radiothérapie. Les objectifs du traitement sont de contrôler la taille de la tumeur, de ramener les taux de GH et d’IGF-I à la normale, d’améliorer les symptômes et de gérer les problèmes de santé associés. Aucun traitement ne convient à tout le monde. Votre médecin vous recommandera un plan de traitement qui vous convient, en fonction de facteurs tels que votre âge, la taille de la tumeur, la gravité des symptômes, les taux de GH et d’IGF-I et l’état de santé.
Chirurgie
Les médecins peuvent retirer la plupart des tumeurs hypophysaires en utilisant une méthode appelée chirurgie transsphénoïdale. L’opération se fait par le nez et le sinus sphénoïdal, un espace creux dans le crâne derrière les voies nasales et sous le cerveau. Deux approches pour cette chirurgie sont
- avec un microscope – un outil grossissant
- avec un endoscope – un tube fin et éclairé avec une petite caméra
Dans les deux approches, le chirurgien utilise une imagerie IRM avancée pour scanner la zone autour de la tumeur avant la chirurgie. Il ou elle fait ensuite une petite incision à l’intérieur de votre narine pour voir la zone et enlever la tumeur à l’aide de minuscules outils spéciaux. En chirurgie microscopique, le chirurgien utilise un microscope pour agrandir la zone. En chirurgie endoscopique, une caméra endoscopique envoie des images à un moniteur de télévision à la place. Les risques et les résultats sont similaires pour les deux approches.3
Lorsque la tumeur qui crée trop de GH n’est pas localisée dans l’hypophyse, d’autres types de chirurgie sont utilisés pour enlever la tumeur. L’élimination de ces tumeurs non pituitaires abaisse également les niveaux de GH et améliore les symptômes d’acromégalie.
Risques. Les complications de la chirurgie peuvent inclure des saignements, des fuites de liquide céphalo-rachidien, une méningite, un déséquilibre sodique (sel) et hydrique et de faibles niveaux d’hormones hypophysaires.3
Résultats.La chirurgie est considérée comme un succès si les taux sanguins de GH et d’IGF-I reviennent à la normale après 12 semaines. Le taux de guérison juste après la chirurgie est d’environ 85% pour les petites tumeurs et de 40 à 50% pour les grosses tumeurs.3
En cas de succès, la chirurgie soulage la pression sur les zones voisines du cerveau et fait chuter les niveaux de GH vers la droite. une façon. L’enflure des tissus mous peut s’améliorer en quelques jours, mais les changements du visage peuvent prendre plus de temps à s’améliorer.
La chirurgie est plus efficace chez les personnes atteintes de petites tumeurs hypophysaires. Le succès dépend en grande partie des compétences et de l’expérience du chirurgien, ainsi que de l’emplacement de la tumeur. Même les chirurgiens expérimentés peuvent ne pas être en mesure de retirer la tumeur si elle est trop proche de parties du cerveau où la chirurgie serait risquée. Cependant, les chirurgiens peuvent être en mesure de retirer une partie de la tumeur.
Traitements post-chirurgicaux. Dans la plupart des cas, les niveaux de GH et d’IGF-I s’améliorent mais ne reviennent pas à la normale. Si les niveaux de ces hormones sont encore trop élevés ou recommencent à augmenter, un traitement supplémentaire peut être nécessaire. Le plus souvent, cela impliquera la prise de médicaments. Dans certains cas, votre médecin peut recommander une deuxième intervention chirurgicale.
Médicaments
Actuellement, trois types de médicaments sont utilisés pour traiter l’acromégalie, mais ils ne permettent pas de guérir. Les médicaments peuvent être utilisés seuls ou en association les uns avec les autres.
Analogues de la somatostatine. Les médicaments les plus souvent utilisés pour traiter l’acromégalie sont appelés analogues de la somatostatine (SSA). Ces médicaments freinent la libération de GH et peuvent également réduire la taille de la tumeur hypophysaire. Plusieurs études ont montré que ces médicaments sont sûrs et efficaces pour un traitement à long terme. Les médicaments sont administrés par injection, mais les scientifiques étudient actuellement d’autres options, telles que les pilules.4 Les effets secondaires les plus courants des SSA sont les crampes, les gaz et la diarrhée. Ces effets sont généralement légers et disparaissent avec le temps. Certaines personnes peuvent développer des calculs biliaires qui ne provoquent généralement pas de symptômes. La chute des cheveux est possible et, dans de rares cas, permanente. Le contrôle de la glycémie s’améliore généralement mais, rarement, peut s’aggraver.
Agonistes de la dopamine. Ces médicaments inhibent la production de GH et la croissance tumorale, mais pas aussi bien que les SSA. Les agonistes de la dopamine sont plus susceptibles d’agir chez les personnes qui ont un léger excès de GH et celles qui ont à la fois une acromégalie et une hyperprolactinémie (trop d’hormone prolactine). Les médicaments sont pris par voie orale. Les effets secondaires peuvent inclure des nausées, un nez bouché, de la fatigue, des maux de tête, des étourdissements en position debout, des cauchemars et des changements d’humeur.
Antagonistes des récepteurs de l’hormone de croissance. Contrairement aux deux autres médicaments, les antagonistes des récepteurs GH n’empêchent pas le corps de produire trop de GH. Au lieu de cela, ils empêchent GH de signaler au corps de produire plus d’IGF-I. Le médicament est pris sous la forme d’une injection quotidienne sous la peau que les patients peuvent s’administrer eux-mêmes. Les effets secondaires peuvent inclure des problèmes hépatiques.
La radiothérapie
La troisième option de traitement est la radiothérapie, qui utilise des rayons X à haute énergie ou des ondes de particules pour tuer les cellules tumorales. Ce type de traitement peut être recommandé si la chirurgie n’est pas possible ou ne parvient pas à retirer tous les tissus tumoraux, et que les médicaments ne sont pas une option ou ne fonctionnent pas pour vous.
Stéréotaxique. Le type de radiothérapie préféré est la radiothérapie stéréotaxique, qui utilise l’imagerie 3D pour cibler avec précision de fortes doses de rayonnement sur la tumeur sous différents angles.3 Le traitement peut parfois être effectué en une seule séance, ce qui réduit le risque de dommages à proximité. tissu. Cependant, une dose unique peut ne pas fonctionner pour les très grosses tumeurs et les tumeurs situées à proximité des nerfs qui affectent la vision.
Conventionnel. La deuxième option est la radiothérapie conventionnelle, qui cible également la tumeur avec des faisceaux externes. Ce type de radiothérapie délivre de petites doses de rayonnement dans une série de traitements sur 4 à 6 semaines.
La radiothérapie abaisse les niveaux de GH et d’IGF-I au fil du temps, cela peut prendre des années pour que ce traitement améliore sensiblement les symptômes d’acromégalie. Votre médecin vous prescrira probablement des médicaments pendant que vous attendez que les taux de GH et d’IGF-I reviennent à la normale et que les symptômes s’améliorent.
Toutes les formes de radiothérapie entraînent une diminution lente des autres hormones hypophysaires avec le temps. Environ la moitié des personnes traitées par radiothérapie auront besoin d’un remplacement hormonal après la fin du traitement. Les radiations peuvent également altérer la fertilité d’un patient.
La perte de vision et les lésions cérébrales sont des complications rares. Rarement, d’autres types de tumeurs peuvent se développer plusieurs années plus tard dans des zones qui étaient sur le chemin du faisceau de rayonnement.
Essais cliniques pour l’acromégalie
Le NIDDK mène et soutient des essais cliniques dans de nombreux maladies et affections, y compris les maladies endocriniennes. Les essais visent à trouver de nouvelles façons de prévenir, de détecter ou de traiter la maladie et d’améliorer la qualité de vie.
Que sont les essais cliniques sur l’acromégalie?
Les essais cliniques – et d’autres types d’études cliniques – font partie de la recherche médicale et impliquent des personnes comme vous. Lorsque vous vous portez volontaire pour participer à une étude clinique, vous aidez les médecins et les chercheurs à en apprendre davantage sur la maladie et à améliorer les soins de santé pour les personnes à l’avenir.
Les chercheurs étudient de nombreux aspects de l’acromégalie et du gigantisme, tels que
- utilisation de médicaments pour traiter le gigantisme chez les enfants et les adolescents
- facteurs génétiques susceptibles de provoquer le développement de tumeurs hypophysaires, et comment traiter les tumeurs et les complications associées chez les enfants et les adultes
Découvrez si les études cliniques vous conviennent.
Quelles études cliniques sur l’acromégalie recherchent des participants?
Vous pouvez afficher une liste filtrée des études cliniques sur l’acromégalie ouvertes et en cours de recrutement sur
www.ClinicalTrials.gov. Vous pouvez élargir ou réduire la liste pour inclure des études cliniques de l’industrie, des universités et des particuliers; cependant, le NIH n’examine pas ces études et ne peut pas garantir qu’elles sont sûres. Parlez toujours à votre fournisseur de soins de santé avant de participer à une étude clinique.