Abaissement de la pression de vapeur
Comme décrit dans le chapitre sur les liquides et les solides, la pression de vapeur d’équilibre d’un liquide est la pression exercée par son gaz phase lorsque la vaporisation et la condensation se produisent à des vitesses égales:
\ text {liquid} \ rightleftharpoons \ text {gas}
Dissolution d’une substance non volatile dans un liquide volatil entraîne une baisse de la pression de vapeur du liquide. Ce phénomène peut être rationalisé en considérant l’effet des molécules de soluté ajoutées sur les processus de vaporisation et de condensation du liquide. Pour se vaporiser, des molécules de solvant doivent être présentes à la surface de la solution. La présence de soluté diminue la surface disponible pour les molécules de solvant et réduit ainsi la vitesse de vaporisation du solvant. Étant donné que le taux de condensation n’est pas affecté par la présence de soluté, le résultat net est que l’équilibre de vaporisation-condensation est atteint avec moins de molécules de solvant dans la phase vapeur (c’est-à-dire à une pression de vapeur inférieure) (Figure 1). Bien que cette interprétation cinétique soit utile, elle ne tient pas compte de plusieurs aspects importants de la nature colligative de l’abaissement de la pression de vapeur. Une explication plus rigoureuse implique la propriété de l’entropie, un sujet de discussion dans un chapitre de texte ultérieur sur la thermodynamique. Pour comprendre l’abaissement de la pression de vapeur d’un liquide, il convient de noter que la plus grande entropie d’une solution par rapport à son solvant et son soluté séparés sert à stabiliser efficacement les molécules de solvant et à empêcher leur vaporisation. Une pression de vapeur plus faible en résulte, et un point d’ébullition proportionnellement plus élevé, comme décrit dans la section suivante de ce module.
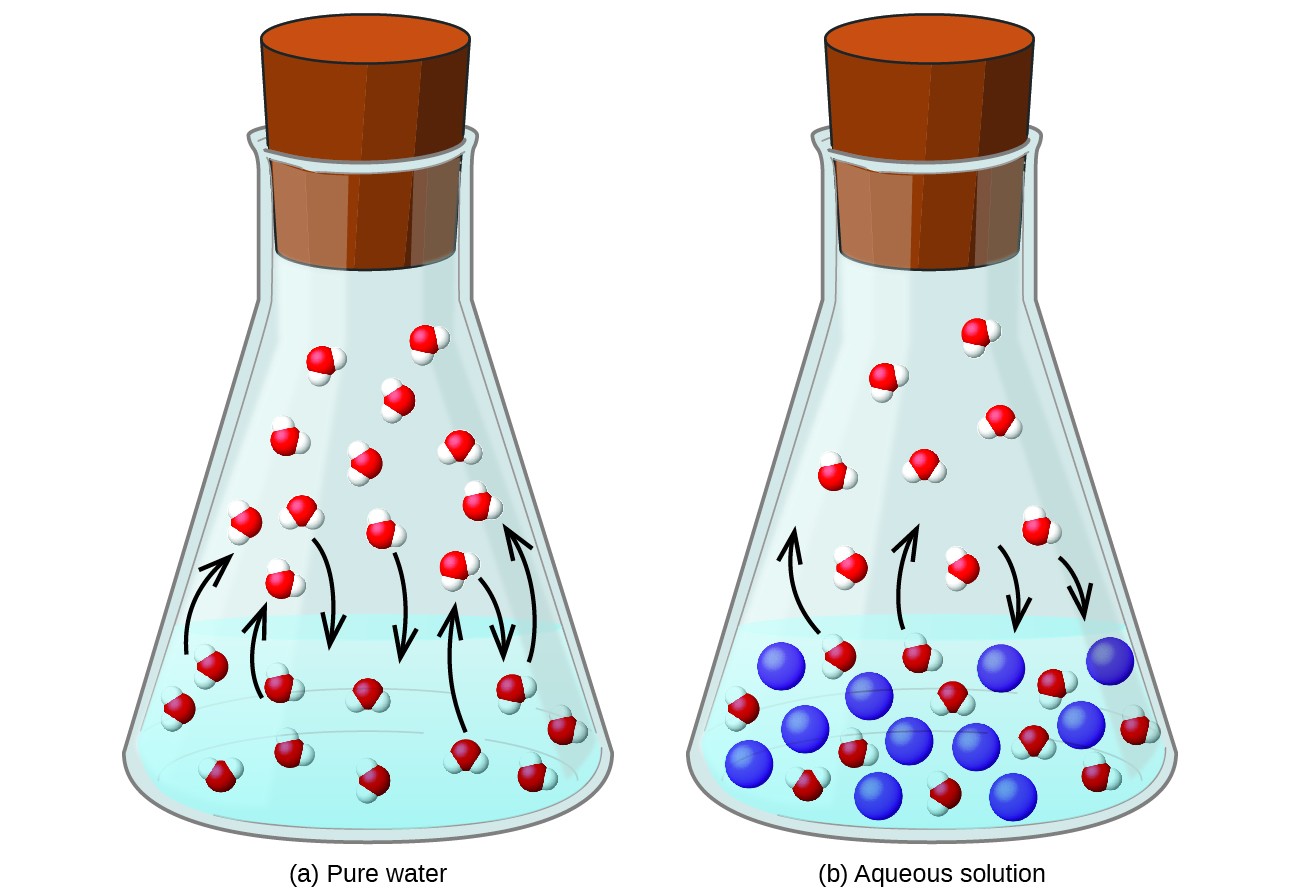
Figure 1. La présence de solutés non volatils abaisse la pression de vapeur d’une solution en empêchant l’évaporation des molécules de solvant.
La relation entre les pressions de vapeur des composants de la solution et les concentrations de ces composants sont décrits par la loi de Raoult: la pression partielle exercée par tout composant d’une solution idéale est égale à la pression de vapeur du composant pur multipliée par sa fraction molaire dans la solution.
{P} _ {\ text {A}} = {X} _ {\ text {A}} {P} _ {\ text {A}} ^ {\ star}
En rappelant que la pression totale d’un mélange gazeux est égale à la somme des pressions partielles pour tous ses composants (loi de Dalton des pressions partielles), la pression de vapeur totale exercée par une solution contenant i composants est
{ P} _ {\ text {solution}} = \ sum _ {i} {P} _ {i} = \ sum _ {i} {X} _ {i} {P} _ {i} ^ {\ star}
Une substance non volatile est une substance dont la pression de vapeur est négligeable (P ^ {\ star} ≈ 0), et donc la pression de vapeur au-dessus d’une solution ne contenant que des solutés non volatils est due uniquement au solvant:
{P} _ {\ text {solution}} = {X} _ {\ text {solvant}} {P} _ {\ text {solvant}} ^ {\ star}
Distillation des solutions
La distillation est une technique de séparation des composants de mélanges largement appliquée en laboratoire et en milieu industriel. Il est utilisé pour raffiner le pétrole, pour isoler les produits de fermentation et pour purifier l’eau. Cette technique de séparation implique le chauffage contrôlé d’un mélange d’échantillon pour vaporiser, condenser et collecter sélectivement un ou plusieurs composants d’intérêt. Un appareil typique pour les distillations à l’échelle du laboratoire est illustré à la Figure 2.
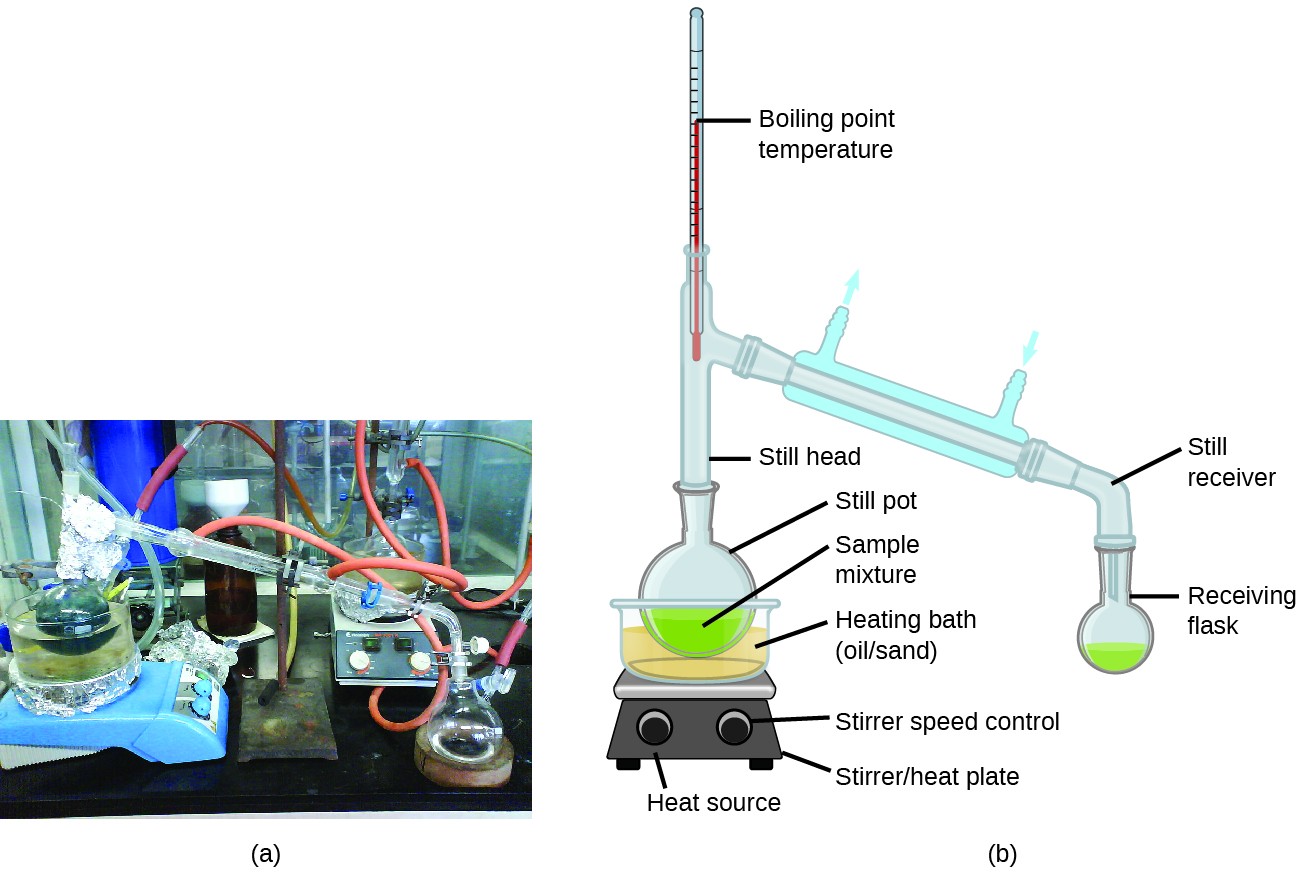
Figure 2. Un modèle typique L’unité de distillation de laboratoire est représentée sur (a) une photographie et (b) un diagramme schématique des composants. (crédit a: modification du travail par « Rifleman82 » / Wikimedia commons; crédit b: modification du travail par « Slashme » / Wikipedia)
Les raffineries de pétrole utilisent une distillation fractionnée à grande échelle pour séparer le composants du pétrole brut. Le pétrole brut est chauffé à des températures élevées à la base d’une haute colonne de fractionnement, vaporisant de nombreux composants qui montent à l’intérieur de la colonne. Lorsque les composants vaporisés atteignent des zones suffisamment froides pendant leur remontée, ils se condensent et sont collectés. Les liquides collectés sont des mélanges plus simples d’hydrocarbures et d’autres composés pétroliers qui sont de composition appropriée pour diverses applications (par exemple, carburant diesel, kérosène, essence), comme le montre la figure 3.
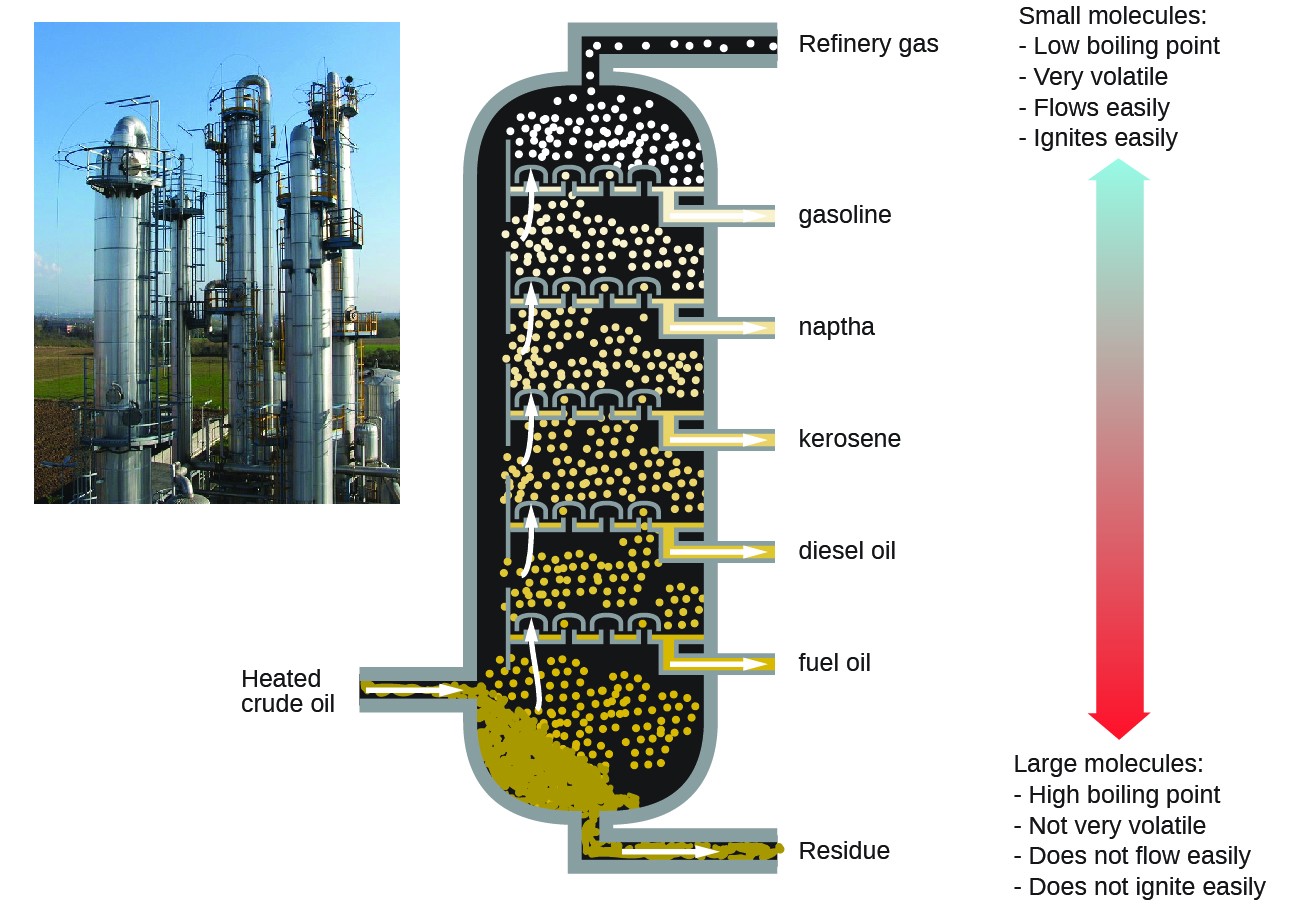
Figure 3. Le pétrole brut est un mélange complexe qui est séparé par distillation fractionnée à grande échelle pour isoler divers mélanges plus simples.
Élévation du point d’ébullition
Comme décrit dans le chapitre sur les liquides et les solides, le point d’ébullition d’un liquide est la température à laquelle sa pression de vapeur est égale à la pression atmosphérique ambiante.Étant donné que la pression de vapeur d’une solution est abaissée en raison de la présence de solutés non volatils, il va de soi que le point d’ébullition de la solution sera ultérieurement augmenté. Par rapport au solvant pur, une solution nécessitera donc une température plus élevée pour atteindre une pression de vapeur donnée, y compris une pression équivalente à celle de l’atmosphère environnante. L’augmentation du point d’ébullition observée lorsque le soluté non volatil est dissous dans un solvant, ΔTb, est appelée élévation du point d’ébullition et est directement proportionnelle à la concentration molaire des espèces de soluté:
\ Delta {T} _ {\ text {b}} = {K} _ {\ text {b}} m
où Kb est la constante d’élévation du point d’ébullition, ou la constante ébullioscopique et m est le concentration molaire (molalité) de toutes les espèces de soluté.
Les constantes d’élévation du point d’ébullition sont des propriétés caractéristiques qui dépendent de l’identité du solvant. Les valeurs de Kb pour plusieurs solvants sont énumérées dans le tableau 1.
La mesure dans laquelle la pression de vapeur d’un solvant est abaissée et le point d’ébullition est élevé dépend du nombre total de particules de soluté présentes dans une quantité donnée de solvant, pas sur la masse, la taille ou l’identité chimique des particules. Une solution aqueuse de 1 m de saccharose (342 g / mol) et une solution aqueuse de 1 m d’éthylène glycol (62 g / mol) présenteront le même point d’ébullition car chaque solution contient une mole de particules de soluté (molécules) par kilogramme de solvant .
Exemple 6: Le point d’ébullition d’une solution d’iode
Trouvez le point d’ébullition d’une solution de 92,1 g d’iode, I2, dans 800,0 g de chloroforme, CHCl3 , en supposant que l’iode est non volatil et que la solution est idéale.

Vérifiez votre apprentissage
Quel est le point d’ébullition d’une solution de 1,0 g de glycérine, C3H5 (OH) 3, dans 47,8 g d’eau? Supposons une solution idéale.
Dépression du point de congélation

Figure 4. Sel gemme ( NaCl), du chlorure de calcium (CaCl2) ou un mélange des deux sont utilisés pour faire fondre la glace. (crédit: modification du travail par Eddie Welker)
Les solutions gèlent à des températures plus basses que les liquides purs. Ce phénomène est exploité dans des schémas de «dégivrage» qui utilisent du sel (figure 4), du chlorure de calcium ou de l’urée pour faire fondre la glace sur les routes et les trottoirs, et dans l’utilisation de l’éthylène glycol comme «antigel» dans les radiateurs d’automobiles. L’eau de mer gèle à une température plus basse que l’eau douce, de sorte que les océans Arctique et Antarctique restent non gelés même à des températures inférieures à 0 ° C (tout comme les fluides corporels des poissons et autres animaux marins à sang froid qui vivent dans ces océans).
La diminution du point de congélation d’une solution diluée par rapport à celle du solvant pur, ΔTf, est appelée dépression du point de congélation et est directement proportionnelle à la concentration molaire du soluté
\ Delta {T} _ {\ text {f}} = {K} _ {\ text {f}} m
où m est la concentration molaire du soluté dans le solvant et Kf sont appelés la constante de dépression du point de congélation (ou constante cryoscopique). Tout comme pour les constantes d’élévation du point d’ébullition, ce sont des propriétés caractéristiques dont les valeurs dépendent de l’identité chimique du solvant. Les valeurs de Kf pour plusieurs solvants sont énumérées dans le tableau 1.
Propriétés colligatives et dégivrage
Le chlorure de sodium et ses analogues du groupe 2, le calcium et le chlorure de magnésium sont souvent utilisés pour déglacer les chaussées et les trottoirs, en raison du fait qu’une solution de l’un de ces sels aura un point de congélation inférieur à 0 ° C, le point de congélation de l’eau pure. Les sels métalliques du groupe 2 sont fréquemment mélangés avec le chlorure de sodium («sel gemme») moins cher et plus facilement disponible pour une utilisation sur les routes, car ils ont tendance à être un peu moins corrosifs que le NaCl, et ils fournissent une plus grande dépression du point de congélation , puisqu’ils se dissocient pour donner trois particules par unité de formule, plutôt que deux particules comme le chlorure de sodium.
Parce que ces composés ioniques ont tendance à accélérer la corrosion du métal, ils ne seraient pas un choix judicieux à utiliser dans antigel pour le radiateur de votre voiture ou pour dégivrer un avion avant le décollage.Pour ces applications, des composés covalents, tels que l’éthylène ou le propylène glycol, sont souvent utilisés. Les glycols utilisés dans le liquide de radiateur abaissent non seulement le point de congélation du liquide, mais ils élèvent le point d’ébullition, rendant le fluide utile en hiver et en été. Des glycols chauffés sont souvent pulvérisés sur la surface des avions avant le décollage par mauvais temps en hiver pour enlever la glace qui s’est déjà formée et empêcher la formation de plus de glace, qui serait particulièrement dangereuse si elle se forme sur les gouvernes de l’avion (Figure 5).

Figure 5. La dépression du point de congélation est exploitée pour enlever la glace (a) des routes et (b) les gouvernes des aéronefs.
Diagramme de phase pour une solution
Les effets colligatifs sur la pression de vapeur, le point d’ébullition et le point de congélation décrits dans la section précédente sont convenablement résumés en comparant les diagrammes de phase pour un liquide pur et une solution dérivée de ce liquide. Les diagrammes de phase pour l’eau et une solution aqueuse sont présentés à la Figure 6.
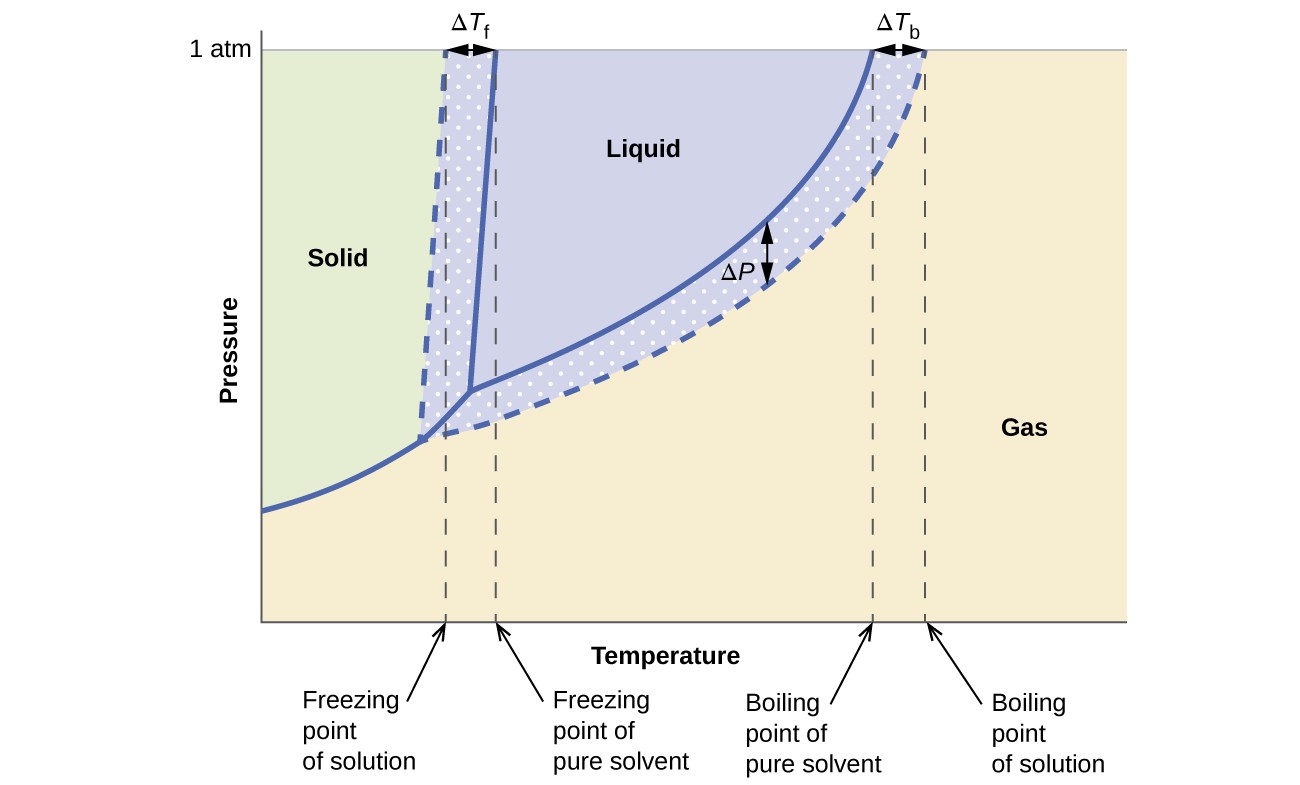
Figure 6. Ces phases les diagrammes montrent l’eau (courbes pleines) et une solution aqueuse de non-électrolyte (courbes en pointillés).
La courbe liquide-vapeur pour la solution est située sous la courbe correspondante pour le solvant, représentant la vapeur abaissement de la pression, ΔP, qui résulte de la dissolution du soluté non volatil. Par conséquent, à toute pression donnée, le point d’ébullition de la solution est observé à une température plus élevée que celle du solvant pur, reflétant l’élévation du point d’ébullition, ΔTb, associée à la présence de soluté non volatil. La courbe solide-liquide de la solution est décalée à gauche de celle du solvant pur, représentant la dépression du point de congélation, ΔTb, qui accompagne la formation de la solution. Enfin, notez que les courbes solide-gaz du solvant et de sa solution sont identiques. C’est le cas de nombreuses solutions comprenant des solvants liquides et des solutés non volatils. Tout comme pour la vaporisation, lorsqu’une solution de ce type est congelée, ce ne sont en fait que les molécules de solvant qui subissent la transition liquide-solide, formant un solvant solide pur qui exclut les espèces de soluté. Les phases solide et gazeuse, par conséquent, sont composées uniquement de solvant, et les transitions entre ces phases ne sont donc pas soumises à des effets colligatifs.
Osmose et pression osmotique des solutions
Un certain nombre de et les matériaux synthétiques présentent une perméation sélective, ce qui signifie que seules des molécules ou des ions d’une certaine taille, forme, polarité, charge, etc., sont capables de traverser (imprégner) le matériau. Les membranes cellulaires biologiques fournissent des exemples élégants de perméation sélective dans la nature, tandis que la tubulure de dialyse utilisée pour éliminer les déchets métaboliques du sang est un exemple technologique plus simpliste. Indépendamment de la façon dont ils peuvent être fabriqués, ces matériaux sont généralement appelés membranes semi-perméables.
Considérez l’appareil illustré à la figure 7, dans lequel des échantillons de solvant pur et une solution sont séparés par une membrane qui ne dissout que les molécules peuvent pénétrer. Les molécules de solvant diffuseront à travers la membrane dans les deux sens. Etant donné que la concentration de solvant est plus élevée dans le solvant pur que dans la solution, ces molécules diffuseront du côté solvant de la membrane vers le côté solution à une vitesse plus rapide qu’elles ne le feront dans le sens inverse. Le résultat est un transfert net de molécules de solvant du solvant pur vers la solution. Le transfert par diffusion de molécules de solvant à travers une membrane semi-perméable est un processus appelé osmose.
Figure 7. a) Une solution et un solvant pur sont initialement séparés par une membrane osmotique. (b) Le transfert net des molécules de solvant vers la solution se produit jusqu’à ce que sa pression osmotique donne des taux de transfert égaux dans les deux sens.
Lorsque l’osmose est effectuée dans un appareil comme celui illustré à la figure 7 , le volume de la solution augmente au fur et à mesure qu’elle se dilue par accumulation de solvant. Cela provoque une augmentation du niveau de la solution, augmentant sa pression hydrostatique (en raison du poids de la colonne de solution dans le tube) et entraînant un transfert plus rapide des molécules de solvant vers le côté du solvant pur. Lorsque la pression atteint une valeur qui donne un taux de transfert de solvant inverse égal au taux d’osmose, le transfert en masse de solvant cesse. Cette pression est appelée la pression osmotique (Π) de la solution. La pression osmotique d’une solution diluée est liée à sa molarité de soluté, M, et sa température absolue, T, selon l’équation
\ Pi = MRT
où R est la constante de gaz universelle.
Si une solution est placée dans un appareil comme celui illustré à la figure 8, l’application d’une pression supérieure à la pression osmotique de la solution inverse l’osmose et pousse les molécules de solvant de la solution dans le solvant pur. Cette technique d’osmose inverse est utilisée pour le dessalement à grande échelle de l’eau de mer et à plus petite échelle pour produire de l’eau du robinet de haute pureté à boire.
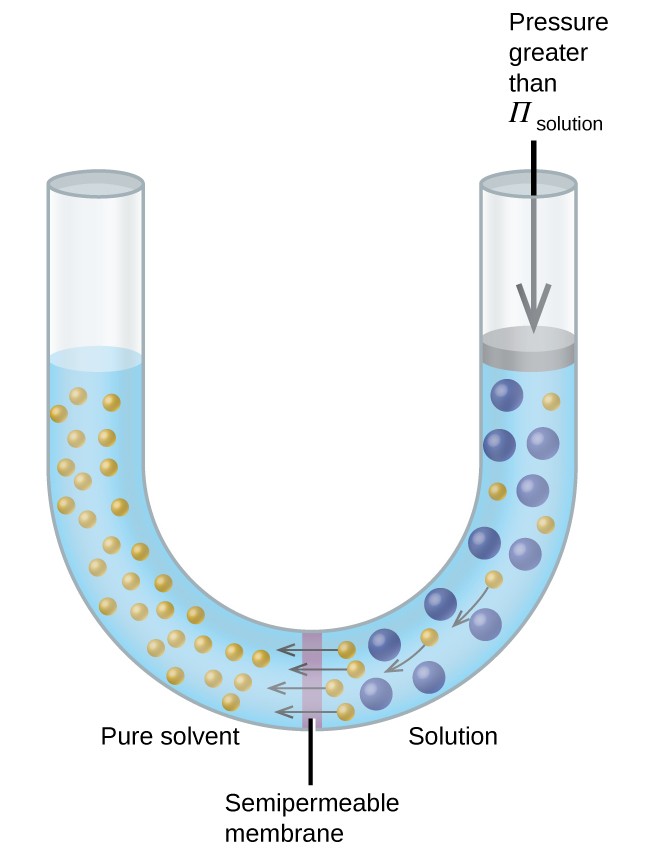
Figure 8. L’application d’une pression supérieure à la pression osmotique d’une solution inversera l’osmose. Les molécules de solvant de la solution sont poussées dans le solvant pur.
Purification de l’eau par osmose inverse
Dans le processus d’osmose, la diffusion sert à déplacer l’eau à travers un membrane semi-perméable d’une solution moins concentrée à une solution plus concentrée. La pression osmotique est la quantité de pression qui doit être appliquée à la solution la plus concentrée pour provoquer l’arrêt de l’osmose. Si une pression plus élevée est appliquée, l’eau passera de la solution la plus concentrée à une solution moins concentrée (plus pure). C’est ce qu’on appelle l’osmose inverse. L’osmose inverse (RO) est utilisée pour purifier l’eau dans de nombreuses applications, des usines de dessalement dans les villes côtières aux machines de purification de l’eau dans les épiceries (figure 9) et aux petites unités ménagères à osmose inverse. Avec une pompe manuelle, de petites unités RO peuvent être utilisées dans les pays du tiers monde, les zones sinistrées et les canots de sauvetage. Nos forces militaires disposent d’une variété d’unités RO fonctionnant avec des générateurs qui peuvent être transportées dans des véhicules vers des endroits éloignés.

Figure 9. Les systèmes d’osmose inverse pour la purification de l’eau potable sont représentés ici à (a) petite et (b) grande échelle. (crédit a: modification du travail de Jerry Kirkhart; crédit b: modification du travail de Willard J. Lathrop)
Des exemples d’osmose sont évidents dans de nombreux systèmes biologiques car les cellules sont entouré de membranes semi-perméables. Les carottes et le céleri qui sont devenus mous parce qu’ils ont perdu de l’eau peuvent à nouveau être croustillants en les plaçant dans l’eau. L’eau pénètre dans les cellules de carotte ou de céleri par osmose. Un concombre placé dans une solution saline concentrée perd de l’eau par osmose et absorbe du sel pour devenir un cornichon. L’osmose peut également affecter les cellules animales. Les concentrations de soluté sont particulièrement importantes lorsque des solutions sont injectées dans le corps. Les solutés dans les fluides cellulaires corporels et le sérum sanguin donnent à ces solutions une pression osmotique d’environ 7,7 atm. Les solutions injectées dans le corps doivent avoir la même pression osmotique que le sérum sanguin; c’est-à-dire qu’ils doivent être isotoniques avec le sérum sanguin. Si une solution moins concentrée, une solution hypotonique, est injectée en quantité suffisante pour diluer le sérum sanguin, l’eau du sérum dilué passe dans les cellules sanguines par osmose, provoquant l’expansion et la rupture des cellules. Ce processus s’appelle l’hémolyse. Lorsqu’une solution plus concentrée, une solution hypertonique, est injectée, les cellules perdent de l’eau en solution plus concentrée, se ratatinent et meurent éventuellement dans un processus appelé crénelage. Ces effets sont illustrés à la Figure 10.
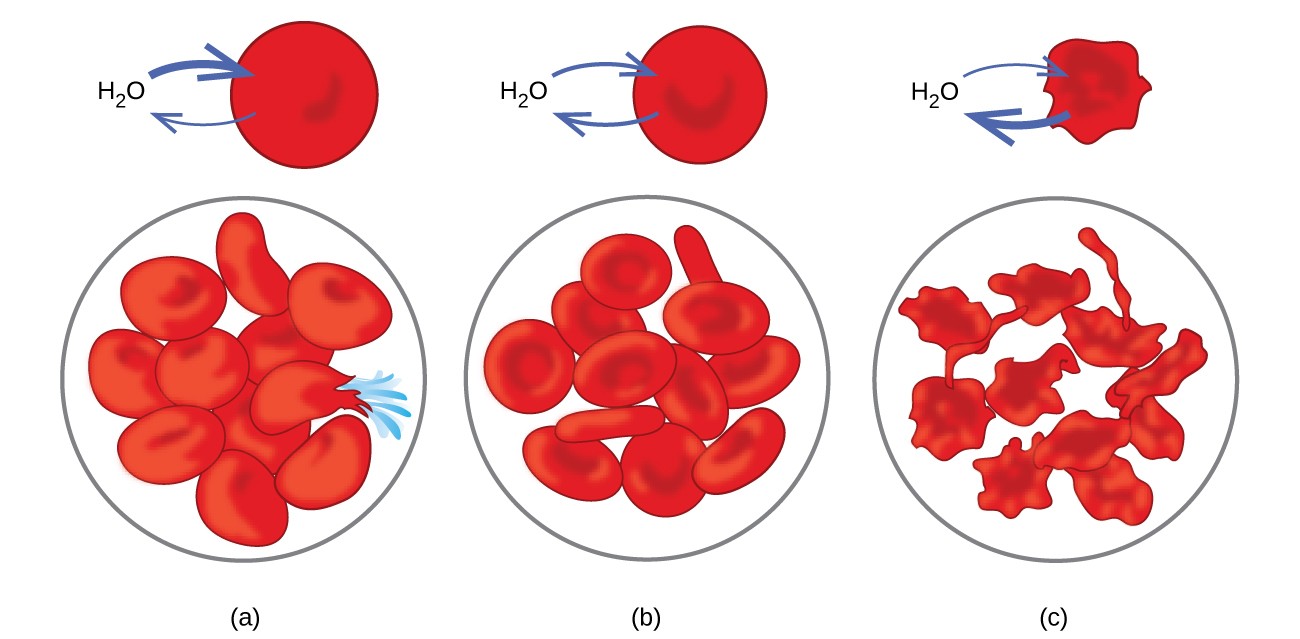
Figure 10. Les membranes des globules rouges sont perméables à l’eau et va (a) gonfler et éventuellement se rompre dans une solution hypotonique; (b) maintenir un volume et une forme normaux dans une solution isotonique; et (c) se ratatiner et éventuellement mourir dans une solution hypertonique. (crédit a / b / c: modifications du travail par « LadyofHats » / Wikimedia commons)
Détermination des masses molaires
Pression osmotique et changements de point de congélation, ébullition et la pression de vapeur sont directement proportionnelles à la concentration de soluté présent. Par conséquent, nous pouvons utiliser une mesure de l’une de ces propriétés pour déterminer la masse molaire du soluté à partir des mesures.
Propriétés colligatives des électrolytes
Comme indiqué précédemment dans ce module, les propriétés colligatives d’une solution dépendent uniquement du nombre, et non de l’identité, des espèces de soluté dissoutes. Les termes de concentration dans les équations pour différentes propriétés colligatives (dépression du point de congélation , élévation du point d’ébullition, pression osmotique) se rapportent à toutes les espèces de soluté présentes dans la solution. Pour les solutions considérées jusqu’ici dans ce chapitre, les solutés étaient des non-électrolytes qui se dissolvent physiquement sans dissociation ou tout autre processus d’accompagnement. Chaque molécu le qui se dissout donne une molécule de soluté dissoute. La dissolution d’un électroyte n’est cependant pas aussi simple, comme l’illustrent les deux exemples courants ci-dessous:
En considérant le premier de ces exemples, et en supposant une dissociation complète, une solution aqueuse de 1,0 m de NaCl contient 2,0 mole d’ions (1,0 mol Na + et 1.0 mol Cl−) pour chaque kilogramme d’eau, et sa dépression au point de congélation devrait être
Lorsque cette solution est réellement préparée et que sa dépression du point de congélation est mesurée, cependant, une valeur de 3,4 ° C est obtenue. Des écarts similaires sont observés pour d’autres composés ioniques, et les différences entre les valeurs de propriété colligative mesurées et attendues deviennent généralement plus importantes à mesure que les concentrations de soluté augmentent. Ces observations suggèrent que les ions de chlorure de sodium (et autres électrolytes forts) ne sont pas complètement dissociés en solution.
Pour en tenir compte et éviter les erreurs accompagnant l’hypothèse de dissociation totale, un paramètre mesuré expérimentalement nommé dans l’honneur du chimiste allemand Jacobus Henricus van’t Hoff, lauréat du prix Nobel. Le facteur de van’t Hoff (i) est défini comme le rapport des particules de soluté en solution au nombre d’unités de formule dissoutes:
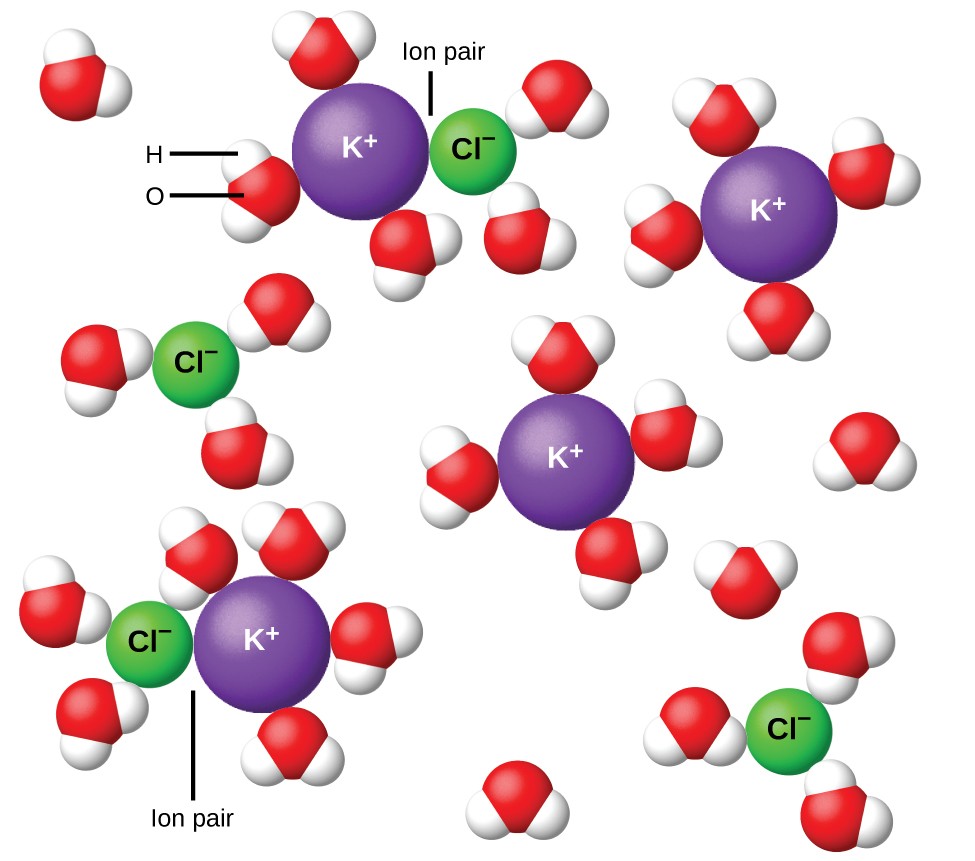
Figure 11. Les ions se séparent de plus en plus largement, plus la solution est diluée, et les attractions interioniques résiduelles diminuent.
En 1923, les chimistes Peter Debye et Erich Hückel ont proposé une théorie pour expliquer l’ionisation incomplète apparente des électrolytes forts. Ils ont suggéré que bien que l’attraction interionique dans une solution aqueuse soit très fortement réduite par la solvatation des ions et l’action isolante du solvant polaire, elle n’est pas complètement annulée. Les attractions résiduelles empêchent les ions de se comporter comme des particules totalement indépendantes (Figure 11). Dans certains cas, un ion positif et négatif peut réellement se toucher, donnant une unité solvatée appelée paire d’ions. Ainsi, l’activité, ou la concentration efficace, de tout type particulier d’ion est inférieure à celle indiquée par la concentration réelle. Les ions se séparent de plus en plus largement, plus la solution est diluée, et les attractions interioniques résiduelles deviennent de moins en moins. Ainsi, dans des solutions extrêmement diluées, les concentrations efficaces des ions (leurs activités) sont essentiellement égales aux concentrations réelles. Notez que les facteurs de van’t Hoff pour les électrolytes du tableau 2 sont pour des solutions de 0,05 m, à quelle concentration la valeur de i pour NaCl est de 1,9, par opposition à une valeur idéale de 2.
Exemple 11: Le point de congélation d’une solution d’électrolyte
La concentration d’ions dans l’eau de mer est sensiblement la même que dans une solution contenant 4,2 g de NaCl dissous dans 125 g d’eau. Supposons que chacun des ions de la solution de NaCl a le même effet sur le point de congélation de l’eau qu’une molécule non électrolytique, et déterminez la température de congélation de la solution (qui est approximativement égale à la température de congélation de l’eau de mer).

Vérifiez votre apprentissage
Supposons que chacun des ions du chlorure de calcium, CaCl2, a le même effet sur le point de congélation de l’eau qu’une molécule non électrolytique. Calculez le point de congélation d’une solution de 0,724 g de CaCl2 dans 175 g d’eau.
Essayer
- La viande peut être classée comme fraîche (non congelée) même si elle est conservée à −1 ° C. Pourquoi la viande ne gèle-t-elle pas à cette température?
- Un composé organique a une composition de 93,46% C et 6,54% H en masse. Une solution de 0,090 g de ce composé dans 1,10 g de camphre fond à 158,4 ° C. Le point de fusion du camphre pur est de 178,4 ° C. Kf pour le camphre est de 37,7 ° C / m. Quelle est la formule moléculaire du soluté? Montrez vos calculs.
- Un sel est connu pour être un fluorure de métal alcalin. Une détermination rapide et approximative du point de congélation indique que 4 g de sel dissous dans 100 g d’eau produisent une solution qui gèle à environ -1,4 ° C. Quelle est la formule du sel? Montrez vos calculs.
Glossaire
activité: concentration efficace d’ions en solution; elle est inférieure à la concentration réelle, en raison des interactions ioniques.
élévation du point d’ébullition: élévation du point d’ébullition d’un liquide par addition d’un soluté
constante d’élévation du point d’ébullition: le constante de proportionnalité dans l’équation reliant l’élévation du point d’ébullition à la molalité du soluté; également connue sous le nom de constante ébullioscopique
propriété colligative: propriété d’une solution qui ne dépend que de la concentration d’une espèce de soluté
crénelage: processus par lequel les cellules biologiques se ratatinent en raison de la perte d’eau par osmose
abaissement du point de congélation: abaissement du point de congélation d’un liquide par ajout d’un soluté
constante de dépression du point de congélation: (aussi, constante cryoscopique) constante de proportionnalité dans l’équation relative dépression du point de congélation pour soluer la molalité
hémolyse: rupture des globules rouges due à l’accumulation d’un excès d’eau par osmose
hypertonique: d’une plus grande pression osmotique
hypotonique : de pression osmotique inférieure
paire d’ions: paire anion / cation solvatée maintenue ensemble par une attraction électrostatique modérée
isotonique: de pression osmotique égale
molalité (m) : une unité de concentration définie comme le rapport du nombre de moles de soluté à la masse du solvant en kilogrammes
fraction molaire (X): le rapport entre la quantité molaire d’un composant de solution et le nombre total de moles de tous les composants de la solution
osmose: diffusion de molécules de solvant à travers une membrane semi-perméable
pression osmotique (Π ): pression opposée nécessaire pour empêcher le transfert massif de molécules de solvant à travers une membrane semi-perméable
Loi de Raoult: la pression partielle exercée par un composant de la solution est égale au produit de la fraction molaire du composant dans la solution et de son équilibre pression de vapeur à l’état pur
membrane semi-perméable: membrane qui permet sélectivement le passage de certains ions ou molécules
van’t Hoff facteur (i): le rapport du nombre de moles de particules dans une solution au nombre de moles d’unités de formule dissoutes dans la solution
- Un non-électrolyte indiqué pour comparaison. ↵