Introduction
Depuis des décennies, les interactions aliments-médicaments (IDE) et herbes-médicaments sont connues pour limiter le succès des traitements médicaux. Le nombre énorme d’interactions possibles entre les variations génétiques, les régimes médicaux et les nombreux composés bioactifs trouvés dans les aliments et les herbes se traduisent par une complexité écrasante. Des outils modernes tels que l’analyse des mégadonnées, l’apprentissage automatique et la simulation des interactions protéine-ligand peuvent nous aider à répondre à toute une série de questions: les choix alimentaires pourraient-ils contribuer à l’échec des régimes thérapeutiques et, si oui, comment? Quels aliments devraient être consommés avant de prendre un médicament prescrit? Et probablement la question la plus excitante: comment pouvons-nous utiliser ces outils pour prédire l’IDE personnel? De toute évidence, de nombreuses réponses résident dans le métabolisme des médicaments, des aliments et des herbes par le cytochrome P450 3A4 (CYP3A4) dans le foie et le tube digestif (Galetin et al., 2010; Basheer et Kerem, 2015).
La majorité des gènes codant pour les enzymes CYP sont polymorphes. À ce jour, la source d’information la plus complète détaillant les allèles du CYP est le Consortium de variation pharmacogène1, dans lequel moins de 100 allèles du CYP3A4 sont représentés. Parmi ceux-ci, moins de 40 sont des SNP exoniques (polymorphismes mononucléotidiques) qui aboutissent à une séquence protéique modifiée. Le petit nombre de sujets dans tous les travaux précédemment publiés sur les mutations du CYP3A4 nous fournit des données limitées concernant les fréquences réelles des mutations du CYP3A4 dans l’ensemble de la population et dans des groupes définis.
Non seulement les informations fiables sur l’incidence des SNP sont incomplètes. , leurs implications cliniques ne sont pas encore claires dans la plupart des cas (Zanger et al., 2014). Comprendre quels SNP peuvent avoir une signification clinique et quand est une tâche extrêmement complexe. Les tests in vitro sont longs, coûteux et pratiquement de faible pertinence compte tenu de la grande quantité de mutations et du nombre infini de combinaisons alimentaires-médicaments. Les méthodes de modélisation moléculaire, y compris les calculs d’amarrage et de liaison d’énergie libre, peuvent servir à prédire les effets potentiels des SNP et de nombreux composés sur le métabolisme médié par le CYP3A4 (Lewis et al., 1998). Par exemple, les interactions non covalentes, hydrophobes, électrostatiques et de van der Waals contribuent toutes à l’orientation d’un composé et donc à sa liaison et à sa réaction sur le site actif d’une enzyme. À leur tour, ceux-ci détermineront l’affinité et la spécificité de l’enzyme pour différents substrats, ainsi que la puissance des inhibiteurs enzymatiques (Kirchmair et al., 2012; Basheer et al., 2017).
Ici, nous proposons un nouveau approche pour mesurer la fréquence allélique des mutations du CYP3A4 dans différents groupes ethniques. Cette approche globale a le pouvoir de mettre en évidence les mutations qui prévalent dans des groupes ethniques particuliers, et combinée au dépistage des substances chimiques en interaction, par exemple les inhibiteurs alimentaires, permettra d’élucider les effets de mutations particulières sur l’interaction médicament-aliment, servant de pas vers la médecine et la nutrition personnalisées. Ce travail peut sensibiliser à l’importance clinique possible des SNP du CYP3A4 altérant les protéines et suggère également quelques outils nécessaires pour la promotion et l’application de la médecine de précision et personnalisée.
Matériel et méthodes
Filtrage de la base de données et analyse des données
Le jeu de données des variantes CYP3A4 a été téléchargé depuis le navigateur gnomAD2 sous forme de fichier CVS. Python 2.7 avec les packages NumPy, pandas et matplotlib a été utilisé pour l’analyse et la visualisation des données (voir la fiche technique supplémentaire S1). La classification hiérarchique agglomérative a été réalisée à l’aide du logiciel Expander 7 (Shamir et al., 2005) avec le coefficient de corrélation de rang Pearson comme mesure des similitudes et du type de liaison complet. Un seuil de distance de 0,6 a été fixé pour le regroupement des SNP.
Modélisation du polymorphisme in silico
La version Maestro 2017-2 (Schrodinger, New York, NY, États-Unis) a été utilisée pour la modélisation informatique. Le modèle d’amarrage CYP3A4 a été construit comme décrit précédemment (Basheer et al., 2017). En bref, la structure cristalline du CYP3A4 (entrée PDB 2V0M) a été traitée, modifiée et affinée en suivant les étapes de l’assistant de préparation des protéines. Une grille d’amarrage avec une contrainte de coordination métallique pour le Fe2 + dans le groupe hème a été générée sur la base du centroïde du kétoconazole dans le site de liaison d’origine dans la structure cristalline. Sept mutations ont été sélectionnées pour les simulations d’amarrage, une en tant que représentant de chaque groupe ethnique (tableaux 1, 2). Pour chaque protéine variante, une mutation ponctuelle unique a été introduite avant les étapes de préparation de la protéine. Les structures 3D de ligands ont été générées à partir de structures 2D de PubChem3 et préparées pour l’amarrage à l’aide de la tâche LigPrep.Le champ de force OPLS3 et les options Glide par défaut pour la précision standard ont été appliqués pour le modèle d’ancrage, à l’exception du fait que la contrainte de coordination métallique a été utilisée, ainsi que 30 poses pour le nombre de poses à inclure et 10 poses pour le nombre de poses à écrire. en dehors. Pour chaque ligand, le résultat d’amarrage avec le score d’émodèle Glide le plus faible a été sélectionné.

Tableau 1. Sélection de SNP représentatifs pour sept groupes ethniques.

Tableau 2. Fréquence (%) des mutations sélectionnées par groupe ethnique.
Résultats
La base de données d’agrégation du génome (gnomAD; voir la note de bas de page 2) regroupe les données de séquençage de l’exome et du génome d’une grande variété de projets de séquençage à grande échelle. Il comprend des données provenant de 125 748 séquences d’exomes et de 15 708 séquences du génome entier de 141 456 individus non apparentés représentant sept populations ethniques (Lek et al., 2016). La base de données GnomAD présente 856 variantes de CYP3A4, dont 397 sont introniques et jusqu’à 459 sont exoniques. Parmi les SNP exoniques, 312 sont des mutations faux-sens, indiquant qu’elles affectent la structure des protéines. Le gène CYP3A4 a une longueur de 34 205 pb. Ses 13 exons comprennent une région codante de 1 512 pb qui produit une protéine de 504 acides aminés. Les 412 SNP exoniques avec des positions uniques dans ce gène donnent une densité de SNP exonique de 272 / kbp (tableau supplémentaire S1).
Le calcul des fréquences d’allèles différentielles par groupe ethnique révèle que certaines populations présentent des fréquences de mutations plus élevées (Figure 1A). La plupart des mutations CYP3A4 dans la population européenne sont en effet rares, comme on le pense généralement, tandis que les mutations dans d’autres populations, telles que l’Afrique et l’Asie de l’Est, sont beaucoup plus fréquentes (tableau supplémentaire S2).
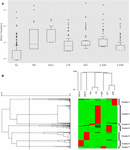
Figure 1. Analyse des SNP faux-sens CYP3A4 dans sept populations distinctes. (A) Boîte à moustaches à échelle logarithmique des fréquences alléliques. Les cases représentent l’intervalle interquartile (IQR), les lignes bleues représentent les médianes, les moustaches représentent les données à moins de 1,5 IQR et les valeurs aberrantes sont représentées par de petits cercles. (B) Regroupement hiérarchique des fréquences alléliques. Chaque ligne représente un seul SNP. Chaque colonne représente une population ethnique distincte. La fréquence allélique des SNP dans chacune des populations est représentée par la couleur de la cellule correspondante dans le fichier matrice. Le vert et le rouge représentent respectivement la basse et la haute fréquence. Le dendrogramme supérieur montre des similitudes dans le modèle de fréquence allélique entre chaque groupe de sujets. Le dendrogramme de gauche représente le regroupement des gènes en deux groupes. La ligne pointillée représente le seuil de distance de 0,6 utilisé pour la division en groupes. UE – Européen (non finlandais; n = 64603), FIN – Européen (finlandais; n = 12,562), ASH J – Juif ashkénaze (n = 5185), LTN – Latino (n = 17,720), AFR – Africain (n = 12 487), E ASN – Asie de l’Est (n = 9 977), S ASN – Asie du Sud (n = 64 603).
Nous avons utilisé le regroupement hiérarchique pour regrouper les variantes avec des modèles de fréquence similaires. Notre analyse des données a produit sept groupes distincts (figure 1B). En outre, il est clairement observé que les SNP à haute fréquence dans chaque cluster sont caractéristiques d’une population spécifique. L’analyse hiérarchique de regroupement des groupes ethniques soutient l’association entre la variance génétique et l’ethnicité en regroupant les ethnies apparentées telles que les Asiatiques du Sud et de l’Est ainsi que les Européens finlandais et non finlandais.
Un modèle de calcul a été utilisé pour évaluer l’influence possible de mutations ponctuelles du CYP3A4 sur sa capacité à se lier aux substrats et aux inhibiteurs. Le CYP3A4 est capable d’oxyder une large gamme de composés endogènes et xénobiotiques. Ici, le kétoconazole a été sélectionné comme médicament représentatif et inhibiteur spécifique très efficace; l’androstènedione et la testostérone ont été sélectionnées comme hormone endogène représentative; et la déméthoxycurcumine et l’épigallocatéchine ont été sélectionnées comme représentants des bioactifs alimentaires. Un modèle d’amarrage a été construit pour prédire les poses de liaison des composés sélectionnés dans le site de liaison CYP3A4. Le modèle a d’abord été validé en restaurant avec succès la pose du kétoconazole dans le site de liaison, avec un RMSD de 1,52 Â par rapport à la structure cristalline d’origine. Sept protéines mutantes ont été conçues sur la base de la structure cristalline de la protéine de type sauvage (figure supplémentaire S1). Pour chaque groupe ethnique, la mutation unique la plus fréquente a été choisie comme représentative. L’effet de mutations uniques sur la liaison au substrat a été évalué sur la base de la comparaison entre les poses d’ancrage sur la protéine native et sur des protéines variantes. Les changements dans les poses d’amarrage en termes de RMSD sont résumés dans le tableau 3.

Tableau 3. RMSD par rapport au WT des ligands d’ancrage aux sites de liaison de sept variants de CYP3A4.
L’effet du SNP CYP3A4 sur la liaison au substrat s’est avéré être spécifique à la mutation-substrat. Dans quelques cas seulement, des mutations ont provoqué une modification de la pose de liaison d’un ligand dans la poche de liaison. La pose d’amarrage de la testostérone était la même dans les sept variantes testées. Les variants E262K, D174H et K168N n’ont pas provoqué de changement de pose de liaison dans aucune des molécules testées. Cependant, les mutations L373F et T163A ont changé la pose de liaison de l’androstènedione afin qu’elle soit positionnée parallèlement au groupe hème plutôt que perpendiculairement à celui-ci, comme dans la protéine WT. De plus, l’androstènedione a été mise en rotation de sorte que le groupe cyclopentanone soit situé à proximité de l’hème, au lieu du groupe cyclohexanone dans la protéine WT. Les mutations S222P et L293P n’ont provoqué qu’une petite rotation dans la pose de liaison de l’androstènedione (figure 2A). De toutes les mutations examinées, seul S222P a provoqué des changements substantiels dans les poses d’amarrage du kétoconazole et de la déméthoxycurcumine au site de liaison (figures 2B, C); alors que pour l’épigallocatéchine, la mutation de changement de pose était L373F (Figure 2D).
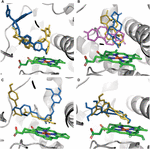
Figure 2. Modèles de ligands ancrés au site de liaison du CYP3A4. (A) Kétoconazole, (B) androstènedione, (C) déméthoxycurcumine et (D) épigallocatéchine. Le site de liaison aux protéines est représenté par des rubans gris; l’hème est représenté par des bâtons verts, les poses d’accostage dans la protéine WT et dans les mutants S222P et L373F sont représentées respectivement par des bâtons orange, bleu et violet. Les poses d’amarrage Androstenedione dans les variantes L293P et T136A chevauchent respectivement les poses des variantes S222P et L373F.
Discussion
Le cytochrome P450 3A4 est la principale enzyme responsable des interactions médicamenteuses. Les recherches actuelles sur les mutations du CYP3A4 se sont concentrées sur quelques dizaines de SNP trouvés dans des études désignées (Sata et al., 2000; Dai et al., 2001; Eiselt et al., 2001; Hsieh et al., 2001; Lamba et al. ., 2002; Murayama et al., 2002). Comme démontré ici, ils représentent la pointe d’un iceberg compte tenu de la prévalence et des résultats potentiels des mutations CYP3A4. L’abondance des projets de séquençage des grands génomes et des exomes a ouvert une nouvelle voie pour l’identification de nombreuses mutations inconnues. Ici, nous montrons que les mutations précédemment présentées ne sont que la pointe de l’iceberg, en mettant en évidence 856 mutations existant dans CYP3A4, dont un tiers modifient la structure protéique. En utilisant une cohorte de 141 456 individus non apparentés, les fréquences alléliques précises des mutations CYP3A4 ont été calculées pour sept ethnies distinctes. À notre connaissance, il s’agit de l’étude à données volumineuses la plus vaste et la plus complète sur les mutations exoniques du CYP3A4 et leurs fréquences alléliques dans différentes populations, publiée à ce jour.
Les enzymes polymorphes CYP3A4 peuvent être très importantes pour expliquer les différences d’efficacité et de toxicité des médicaments entre différents individus. Les mutations du gène CYP3A4 peuvent conduire à une activité enzymatique abolie, réduite, altérée ou accrue. Les mutations exoniques peuvent modifier l’activité enzymatique, comme cela a été démontré dans quelques études cliniques avec des substrats sélectionnés. Certains cas de métabolisme altéré dû aux SNP dans le CYP3A4 ont déjà été décrits dans la littérature (Eiselt et al., 2001; Miyazaki et al., 2008). Malgré l’importance fonctionnelle et la pertinence clinique des SNP dans le CYP3A4 et peut-être en raison de leur fréquence relativement faible identifiée dans la population générale, le polymorphisme dans le CYP3A4 n’a pas reçu l’attention qu’il mérite.
Ici, sept mutations ont servi à prédire l’effet des SNP sur l’orientation de la liaison au substrat et à l’inhibiteur. Dans la littérature, le polymorphisme du CYP3A4 divise la population générale en trois groupes – les métaboliseurs lents, les métaboliseurs normaux et les métaboliseurs rapides, sur la base de SNP introniques qui modifient les niveaux d’expression plutôt que la structure (Zanger et Schwab, 2013). Nos calculs suggèrent une classification supplémentaire: les métaboliseurs modifiés. Certaines mutations proposées par notre modèle virtuel provoqueraient un changement dans l’orientation de liaison des ligands individuels. On s’attend à ce que ces changements réduisent la probabilité d’oxydation enzymatique en raison de l’éloignement accru de l’hème, ou conduisent à des produits qui autrement ne seraient pas évidents au cours des tests de toxicité effectués dans le cadre du processus de développement du médicament. Cependant, comme notre modèle le prédit, pour la plupart des substrats, les mutations du CYP3A4 sont bénignes.
La position modifiée d’un substrat dans la poche de liaison en raison d’un changement structurel de la protéine n’est qu’un mécanisme possible par lequel une mutation pourrait changer l’activité d’une protéine . Un ancrage altéré de la protéine à la membrane, des canaux conducteurs de substrat endommagés et une sortie compromise des produits présentent des mécanismes supplémentaires pour un changement mutationnel dans l’activité d’une protéine. Comme indiqué ici, l’effet de chaque mutation est spécifique au substrat.Déterminer quelles combinaisons de substrats et de mutations pourraient modifier l’activité enzymatique, en utilisant des méthodes in vitro traditionnelles, est laborieux, ce qui souligne la nécessité d’outils virtuels prédictifs pour résoudre ce casse-tête complexe.
Intérêt public et professionnel pour la médecine personnelle et de précision se développe rapidement. La prédiction du métabolisme modifié du médicament sur la base du polymorphisme individuel du CYP3A4 ne semble être qu’une question de temps. Ici, nous proposons que des groupes ethniques distincts portent des ensembles uniques de SNP CYP3A4. En effet, l’ethnicité peut constituer une première étape réalisable en médecine personnalisée, précédant la mise en place d’un dépistage ADN individuel pour tous. Fait intéressant, l’appartenance ethnique a une autre implication pour le métabolisme des médicaments CYP3A4, étant un facteur majeur dans la détermination des choix alimentaires et des habitudes alimentaires. On peut suggérer que les régimes thérapeutiques devraient être spécifiquement conçus pour chaque groupe ethnique, au moins pour les médicaments qui sont fortement métabolisés par le CYP3A4. Cela met en évidence les opportunités d’exploiter et d’intégrer des bases de données et un apprentissage en profondeur pour identifier comment les SNP, l’ethnicité, les composés alimentaires et les médicaments modifient l’activité du CYP3A4 et le succès d’un régime médical.
Disponibilité des données
Des ensembles de données accessibles au public ont été analysés dans cette étude. Ces données peuvent être trouvées ici: http://gnomad.broadinstitute.org/gene/ENSG00000160868.
Contributions des auteurs
Tous les auteurs répertoriés ont apporté une contribution substantielle, directe et intellectuelle à la travail, et l’a approuvé pour publication.
Déclaration de conflit d’intérêts
Les auteurs déclarent que la recherche a été menée en l’absence de toute relation commerciale ou financière qui pourrait être interprétée comme un potentiel conflit d’intérêts.
Matériel supplémentaire
FIGURE S1 | Modèle de ruban 3D du CYP3A4 et localisation des acides aminés mutés dans les sept protéines variantes conçues pour l’amarrage. L’hème est représenté par des bâtonnets verts, Fe2 + est représenté par une sphère rouge, les SNP utilisés dans l’analyse in silico sont représentés par des zones rouges sur le ruban et les groupes R d’acides aminés mutés dans les modèles de variantes sont représentés explicitement par des bâtonnets gris clair.
TABLEAU S1 | Types de SNP CYP3A4 dans une population de 141 456 individus non apparentés représentant 7 populations ethniques.
TABLEAU S2 | SNP CYP3A4 par groupe ethnique.
Notes de bas de page
- ^ www.pharmvar.org
- ^ https://gnomad.broadinstitute.org
- ^ https://pubchem.ncbi.nlm.nih.gov