Le syndrome d’Angelman (SA) est une maladie neurogénétique rare qui affecte environ une personne sur 15 000, soit environ 500 000 personnes dans le monde. Les enfants et les adultes atteints de SA ont généralement des problèmes d’équilibre, une déficience motrice et des convulsions débilitantes. Certaines personnes ne marchent jamais. La plupart ne parlent pas. Les cycles de sommeil perturbés peuvent également être un défi sérieux pour l’individu et le (s) gardien (s). Les personnes atteintes de SA ont besoin de soins continus et sont incapables de vivre de manière autonome. Ils ont une espérance de vie normale. C’est la vie aujourd’hui des personnes atteintes du syndrome d’Angelman, mais l’espoir est là. Les scientifiques pensent que la SA a le plus grand potentiel de guérison par rapport à d’autres troubles neurogénétiques, et FAST (Foundation for Angelman Syndrome Therapeutics) a un plan bien en cours pour y parvenir.
Types Causes Tests & Ressources de diagnostic
Types de syndrome d’Angelman
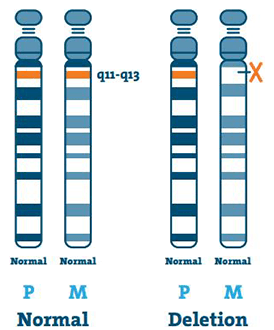
Suppression ( 65-75%)
L’ADN (acide désoxyribonucléique) est le principal composant des chromosomes. Il contient notre code génétique unique. La plupart des personnes atteintes de SA manquent un morceau d’ADN dans la région 15q11-13 sur le chromosome maternel 15.
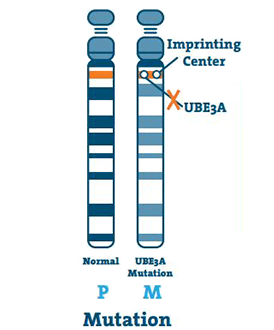
Mutation (5 -11%)
Cela se produit lorsqu’il y a une petite anomalie dans l’ADN du gène UBE3A. Une mutation peut survenir n’importe où sur le gène.
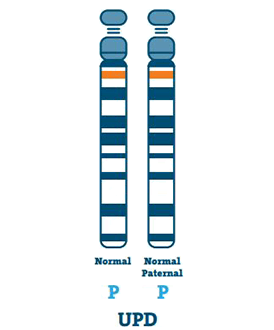
Disomie uniparentale (3-7%)
Un individu avec UPD a deux copies du chromosome 15 de son père, au lieu d’un de chacun du père et de la mère. L’UPD se produit généralement s’il n’y a pas de chromosome 15 dans l’œuf.
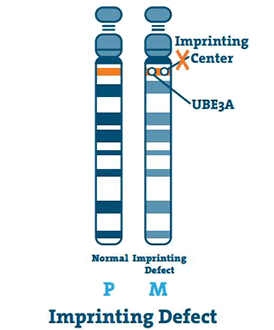
Défaut d’impression (< 3%)
Le centre d’impression est un petit segment d’ADN situé dans la région q11-13 du chromosome. Dans de rares cas, le chromosome 15 de la mère est vierge et le centre copie le chromosome du père 15. Il s’agit d’un défaut d’impression.
Caractéristiques
Les caractéristiques typiques de la SA ne sont généralement pas évidentes à naissance. Les personnes atteintes de ce trouble ont des difficultés d’alimentation pendant l’enfance et un retard de développement notable vers l’âge de 6 à 12 mois. Ils ont besoin de thérapies intensives pour développer leurs compétences fonctionnelles. L’AS affecte toutes les races et les deux sexes. Il est souvent diagnostiqué à tort comme de l’autisme ou de la paralysie cérébrale.
Traits comportementaux
Les personnes atteintes du syndrome d’Angelman ont des traits de comportement distincts, y compris un comportement joyeux, caractérisé par des rires, des sourires et de l’excitabilité fréquents. De nombreuses personnes atteintes de SA ont une fascination pour l’eau et prennent beaucoup de plaisir dans des activités comme la natation et le bain.
Causes du syndrome d’Angelman
Le syndrome d’Angelman est un trouble monogénétique causé par une perte de la fonction du gène UBE3A sur le 15e chromosome maternel. Les gens ont deux ensembles de chromosomes – l’un hérité de la mère et l’autre du père. Chez une personne typique, le gène UBE3A hérité de la mère est actif, tandis que la copie du gène hérité du père est réduite au silence dans les neurones de notre cerveau – un phénomène connu sous le nom d’empreinte. Pour les personnes atteintes de SA, ce gène maternel ne fait pas son travail, et cela a un impact sur leur ARN messager (ARNm).
![]()
Qu’est-ce que l’ARNm?
L’ARNm est le FedEx du corps. Notre ADN utilise l’ARNm comme service de livraison pour envoyer des plans aux usines d’assemblage de protéines de nos cellules. Les personnes atteintes de SA ont une mutation, une suppression ou un autre défaut dans leur gène UBE3A qui interrompt ce service de livraison. En conséquence, leurs neurones ne produisent aucune protéine UBE3A fonctionnelle, et c’est ce qui déclenche les symptômes de la SA. Cette protéine nous aide à marcher, à parler et à effectuer d’autres tâches quotidiennes.
![]()
Est-ce génétique?
Dans la plupart des cas, le syndrome d’Angelman n’est pas héréditaire – en particulier ceux causés par une suppression ou une UPD. Au lieu de cela, ces changements génétiques se produisent comme des événements aléatoires pendant la formation des cellules reproductrices ou au début du développement embryonnaire.
Tests & Diagnostic
En savoir sur les critères que les médecins utilisent pour déterminer si une personne est atteinte du syndrome d’Angelman, et lisez les tests disponibles et les erreurs de diagnostic courantes.
![]()
Test
![]()
Diagnostic

Rejoignez notre communauté
Connectez-vous avec d’autres personnes qui ont un enfant ou un être cher atteint du syndrome d’Angelman.
