Interpretieren von IR-Spektren in 1 Minute oder weniger: Die 2 wichtigsten Dinge, nach denen gesucht werden muss
Im letzten Beitrag haben wir kurz das Konzept der Bindungsschwingungen vorgestellt und festgestellt, dass wir uns kovalente Bindungen ein bisschen wie Kugeln und Federn vorstellen können: Die Federn vibrieren und jede „singt“ mit einer charakteristischen Frequenz Dies hängt von der Stärke der Bindung und von den Massen der Atome ab. Diese Schwingungen haben Frequenzen, die im mittleren Infrarotbereich (IR) des elektromagnetischen Spektrums liegen.
Wir können dies beobachten und messen „Singen“ von Bindungen durch Anlegen von IR-Strahlung an eine Probe und Messen der Frequenzen, bei denen die Strahlung absorbiert wird. Das Ergebnis ist eine als Infrarotspektroskopie bekannte Technik, die ein nützliches und schnelles Werkzeug zur Identifizierung der in einem bestimmten Molekül vorhandenen Bindungen darstellt.
Wir haben gesehen, dass das IR-Spektrum von Wasser ziemlich einfach war – aber weiter zu Als relativ komplexes Molekül wie Glukose (unten) wurden wir plötzlich mit einem Wald von Gipfeln konfrontiert!
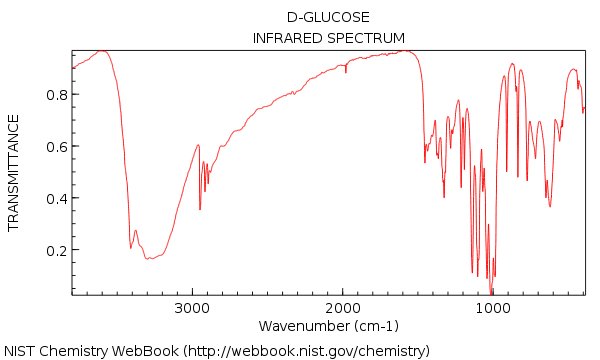
Ihr erster Eindruck vom Schauen an diesem IR könnte sein: agh! Wie soll ich das verstehen?
Zu dem möchte ich sagen: Keine Panik!
Inhaltsverzeichnis
- Korrigieren wir einige häufige Missverständnisse über IR
- Beginnen mit „Hunt and Peck“ ist nicht der richtige Weg
- IR-Spektroskopie: Das große Ganze
- Die beiden wichtigsten Dinge, nach denen in einem IR-Spektrum gesucht werden muss: „Zungen“ und „Schwerter“.
- Alkohole und Carbonsäuren: Mehr Detail
- Spezifische Beispiele für IR-Spektren von Carbonyl-funktionellen Gruppen
- Weniger entscheidend, aber dennoch nützlich: Zwei weitere sehr diagnostische Bereiche.
- Glucose, überarbeitet: Die 1 Minutenanalyse
Korrigieren wir einige häufige Missverständnisse über IR
In diesem Beitrag möchte ich zeigen, dass eine typische Analyse eines IR-Spektrums viel einfacher ist, als Sie vielleicht denken Wenn Sie erst einmal gelernt haben, wonach Sie suchen müssen, können Sie dies oft in einer Minute oder weniger tun. Warum?
- IR wird im Allgemeinen nicht zur Bestimmung der gesamten Struktur eines unbekannten Moleküls verwendet. Zum Beispiel gibt es keine lebende Person, die das obige IR-Spektrum betrachten und die Struktur von Glu ableiten könnte cose daraus. IR ist ein Werkzeug mit einer sehr spezifischen Verwendung.
- Wir müssen nicht jeden einzelnen Peak analysieren! (Wie wir später sehen werden, ist NMR dafür gedacht: -)) Stattdessen eignet sich IR hervorragend zur Identifizierung bestimmter spezifischer funktioneller Gruppen wie Alkohole und Carbonyle. Auf diese Weise ist es komplementär zu anderen Techniken (wie NMR), die diese Informationen nicht so schnell liefern.
Vor diesem Hintergrund können wir die Analyse eines IR-Spektrums durch Ausschneiden vereinfachen alles außer der tiefsten Frucht.
Sehen Sie diesen Wald von Gipfeln von 500-1400 cm-1? Wir werden sie im Grunde alle ignorieren!
80% der nützlichsten Informationen für unsere Zwecke können durch Betrachten von zwei spezifischen Bereichen des Spektrums erhalten werden: 3200-3400 cm-1 und 1650-1800 cm & supmin; ¹. Wir werden auch sehen, dass es mindestens zwei weitere Regionen eines IR-Spektrums gibt, die einen Blick wert sind, und daher eine Analyse „erster Ordnung“ des IR-Spektrums eines Unbekannten abschließen.
Fazit: Die Der Zweck dieses Beitrags ist es, Ihnen zu zeigen, wie Sie Ihre Zeit bei der Analyse eines IR-Spektrums priorisieren können.
2. Mit „Hunt And Peck“ zu beginnen ist nicht der richtige Weg
Was macht ein typischer neuer Schüler mit einem IR-Spektrum eines Unbekannten (und einem Gefühl zunehmender Panik)?
Sie greifen häufig nach dem ersten Werkzeug, das ihnen gegeben wird, nämlich einer Tabelle mit gemeinsamen Bereichen für IR-Peaks, die ihnen von ihrem Ausbilder gegeben wurden.
Der nächste Schritt in ihrer Analyse besteht darin, das Spektrum von einer Seite zur nächsten zu durchlaufen und zu versuchen, jeden einzelnen Peak mit einer der Zahlen in der Tabelle abzugleichen . Ich weiß das, weil ich genau das getan habe, als ich IR zum ersten Mal gelernt habe. Ich nenne es „jagen und picken“.
Die einzigen Menschen, die als ersten Schritt „jagen und picken“, sind Menschen die keinen Plan haben (dh „Neulinge“).
Wenn Sie also die nächsten Absätze lesen, können Sie sich viel Zeit und Verwirrung sparen.
Das große Ganze
In der IR-Spektroskopie messen wir, wo Moleküle Photonen der IR-Strahlung absorbieren. Die Peaks repräsentieren Bereiche des Spektrums, in denen spezifische Bindungsschwingungen auftreten. Genau wie Federn mit unterschiedlichem Gewicht schwingen sie bei charakteristischen Frequenzen in Abhängigkeit von Masse und Spannung.
Hier ist eine Übersicht über das IR-Fenster von 4000 cm -1 bis 500 cm -1, wobei verschiedene interessierende Bereiche hervorgehoben sind.
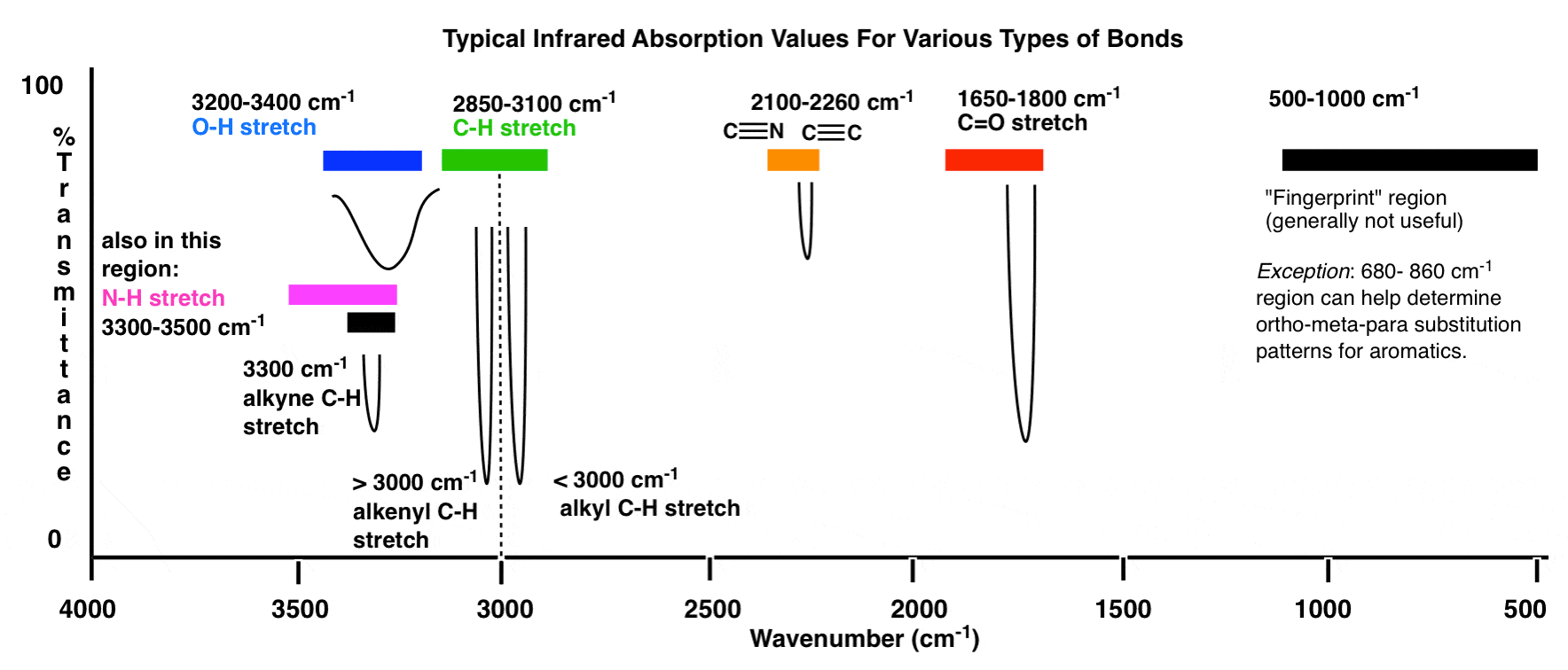
Eine noch komprimiertere Übersicht sieht folgendermaßen aus: (Quelle)
Innerhalb dieser Bereiche gibt es zwei Bereiche mit hoher Priorität, auf die Sie sich konzentrieren müssen, und zwei Bereiche mit niedrigerer Priorität weiter unten diskutieren.
4. Die zwei wichtigsten Dinge, nach denen in einem IR-Spektrum gesucht werden muss: „Zungen“ und „Schwerter“.
Wenn Sie mit einem neuen IR-Spektrum konfrontiert werden, priorisieren Sie Ihre Zeit, indem Sie zwei wichtige Fragen stellen:
- Gibt es in der Region um 3400-3200 cm einen breiten, abgerundeten Peak? 1? Hier treten Hydroxylgruppen (OH) auf.
- Gibt es in der Region um 1850-1630 cm-1 einen scharfen, starken Peak? Hier zeigen sich Carbonylgruppen (C = O).
Schauen wir uns zunächst einige Beispiele für Hydroxylgruppenpeaks in der Region 3400 cm-1 bis 3200 cm-1 an, die Jon beschreibt lebhaft als „Zungen“. Die Peaks unter allen gehören zu Alkoholen. Die Wasserstoffbindung zwischen Hydroxylgruppen führt zu einigen Variationen der OH-Bindungsstärke, was zu einer Reihe von Schwingungsenergien führt. Die Variation führt zu den beobachteten breiten Peaks
Hydroxylgruppen, die Teil von Carbonsäuren sind, haben ein noch breiteres Erscheinungsbild, das wir gleich beschreiben werden.
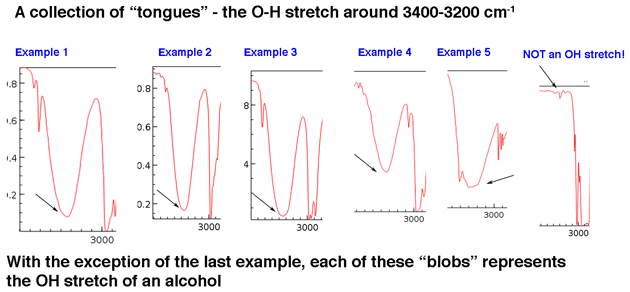
Der Hauptpunkt ist, dass eine Hydroxylgruppe im Grundlinienrauschen im Allgemeinen nicht unbedingt gesucht werden muss.
Obwohl Hydroxylgruppen die häufigste Art von breiten Peaks in dieser Region sind, sind NH-Peaks können auch in diesem Bereich auftreten (mehr dazu in der Fußnote unten). Sie haben tendenziell ein schärferes Erscheinungsbild und können je nach Anzahl der NH-Bindungen als ein oder zwei Peaks auftreten.
Weiter, le Schauen Sie sich einige Beispiele für C = O-Peaks in der Region um 1630-1800 cm-1 an. Diese Peaks sind fast immer die stärksten Peaks im gesamten Spektrum und sind relativ schmal, was ihnen ein etwas „schwertartiges“ Aussehen verleiht
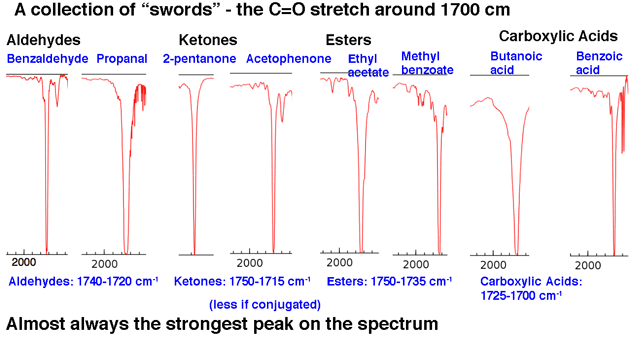
Das fasst unsere 80/20-Analyse zusammen: Suchen Sie nach Zungen und Schwertern.
Wenn Sie lernen nichts anderes aus diesem Beitrag, lernen, diese beiden Arten von Peaks zu erkennen!
Zwei andere Regionen des IR-Spektrums können schnell nützliche Informationen liefern, wenn Sie sich darin üben, nach ihnen zu suchen.
3. Die Linie bei 3000 cm-1 ist eine nützliche „Grenze“ zwischen Alken CH (über 3000 cm-1) und Alkan CH (unter 3000 cm-1). Dies kann Ihnen schnell helfen, festzustellen, ob Doppelbindungen vorhanden sind.
4. Ein Peak in der Region um 2200 cm-1 – 2050 cm-1 ist ein subtiler Indikator für das Vorhandensein einer Dreifachbindung. In dieser Region zeigt sich nichts anderes.
Eine Erinnerung an den gesunden Menschenverstand
Zunächst einige offensichtliche Ratschläge:
- Wenn Sie die Molekülformel erhalten, bestimmt dies, nach welchen funktionellen Gruppen Sie suchen sollten Es ist sinnvoll, nach OH-Gruppen zu suchen, wenn Ihre Molekülformel keinen Sauerstoff enthält, oder auch nach einem Amin, wenn der Formel Stickstoff fehlt.
- Berechnen Sie weniger offensichtlich den Grad der Ungesättigtheit, wenn Sie das Molekül erhalten Formel, weil es wichtige Hinweise liefert. Suchen Sie nicht nach C = O in einer Struktur wie C4H10O, die keinen Grad an Ungesättigtheit aufweist.
5. Alkohole und Carbonsäuren: Weitere Details
Alkohole
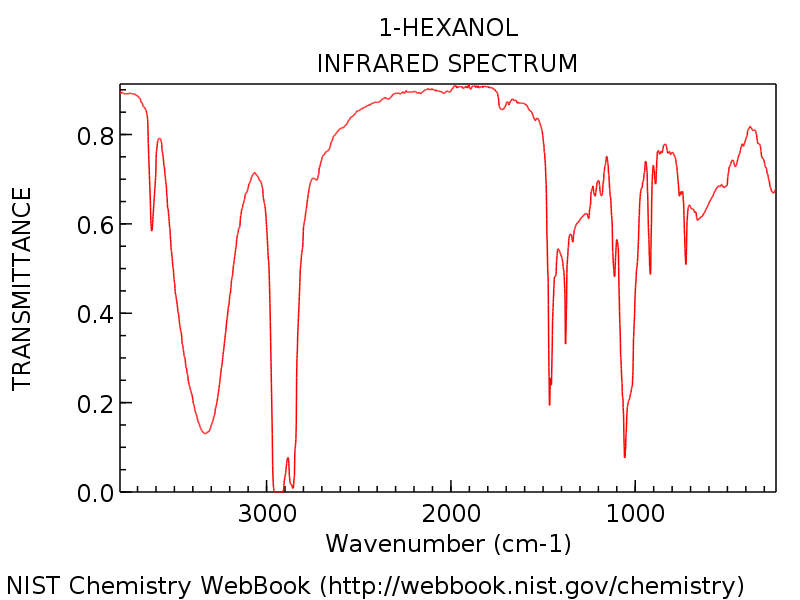
Le Schauen Sie sich ein bestimmtes Beispiel an, damit wir alles in der Perspektive sehen können. Das folgende Spektrum zeigt 1-Hexanol.
Beachten Sie den für einen Alkohol typischen Hydroxylgruppenpeak um 3300 cm & supmin; ¹ (Dieser scharfe Peak um 3600 cm & supmin; ¹ ist ein üblicher Begleiter von Hydroxylpeaks: er repräsentiert nicht wasserstoffgebundenes OH).
Wie Sie es von 1-Hexanol erwarten würden, gibt es keinen verräterischen Carbonylpeak um 1700 cm-1. Anfänger könnten versucht sein, diesen dolchartigen starken Peak bei etwa 1450 cm & supmin; ¹ als mögliche C = O-Dehnung zu bezeichnen. Es ist nicht. (Es ist wahrscheinlich eine C-H-Kurve.) Variationen treten nur in einem sehr engen Bereich auf, und es ist äußerst unwahrscheinlich, dass eine C = O-Dehnung weit unter 1650 cm –1 auftritt. Je mehr Spektren Sie sehen, desto besser können Sie diese Urteile fällen.
Um sich mit Variationen vertraut zu machen, finden Sie hier einige weitere Beispiele von gesamten IR-Spektren verschiedener Alkohole.
- Phenol
- Cyclohexanol
- 1-Butanol
Carbonsäuren
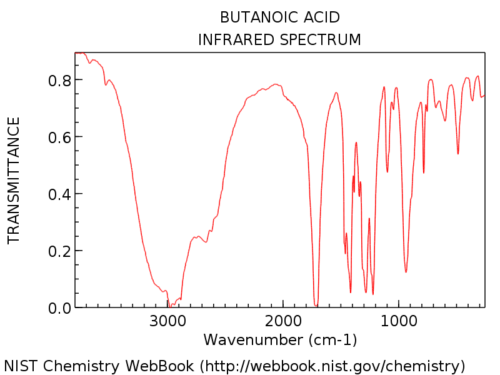
Hydroxylgruppen in Carbonsäuren sind erheblich breiter als in Alkoholen. Jon nennt es einen „haarigen Bart“, was eine perfekte Beschreibung ist. Ihr Aussehen ist ebenfalls sehr variabel. Die OH-Absorption in Carbonsäuren kann so breit sein, dass sie sich unter 3000 cm & supmin; ¹ erstreckt und so ziemlich die linke Hand „übernimmt“ Teil des Spektrums.
Hier ist ein Beispiel: Butansäure.
Hier sind einige weitere Beispiele für vollständige Spektren, damit Sie die Variation sehen können.
- Benzoesäure,
- Pentansäure,
- Essigsäure
Der Unterschied im Aussehen zwischen dem OH eines Alkohols und dem einer Carbonsäure ist normalerweise diagnostisch. In dem seltenen Fall, dass Sie nicht sicher sind, ob der breite Peak auf das OH eines Alkohols oder einer Carbonsäure zurückzuführen ist, sollten Sie den Bereich um 1700 cm auf die C = O-Dehnung überprüfen. Wenn es nicht vorhanden ist, handelt es sich wahrscheinlich um einen Alkohol.
Spezifische Beispiele für IR-Spektren von Carbonylfunktionsgruppen
Die zweite wichtige Peakregion ist die Carbonyl-C = O-Streckfläche bei etwa 1630-1830 cm. Carbonylstrecken sind scharf und stark.
Wenn Sie einige davon sehen, sind sie nicht mehr zu übersehen. In dieser Region wird nichts anderes angezeigt.
Hier ist das IR-Spektrum von Hexanal. Dieser Peak kurz nach 1700 cm & supmin; ¹ ist die C = O-Dehnung. Wenn es vorhanden ist, ist die C = O-Dehnung fast immer der stärkste Peak im IR-Spektrum und nicht zu übersehen.
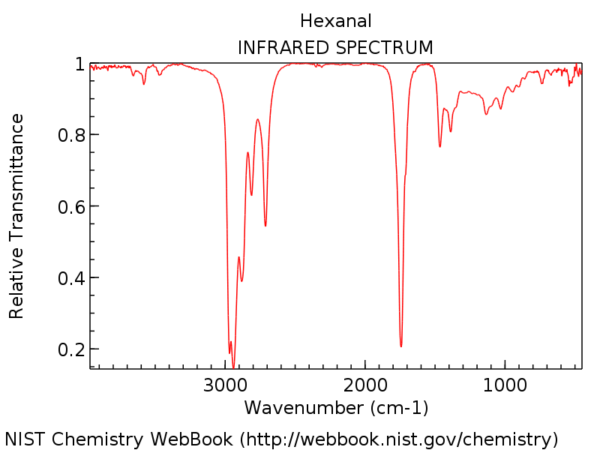
Die Position der C = O-Streckung variiert geringfügig je nach funktioneller Carbonylgruppe. Einige Bereiche (in cm-1) sind nachstehend aufgeführt:
Die Konjugation wirkt sich etwas auf die Position der C = O-Dehnung aus und verschiebt sie auf eine niedrigere Wellenzahl.
Eine anständige Faustregel ist, dass Sie niemals eine C = O-Strecke unter 1630 sehen werden. Wenn Sie beispielsweise bei 1500 einen starken Peak sehen, ist dies nicht C = O. Es ist etwas anderes.
Weniger entscheidend, aber dennoch nützlich: Zwei weitere sehr diagnostische Bereiche.
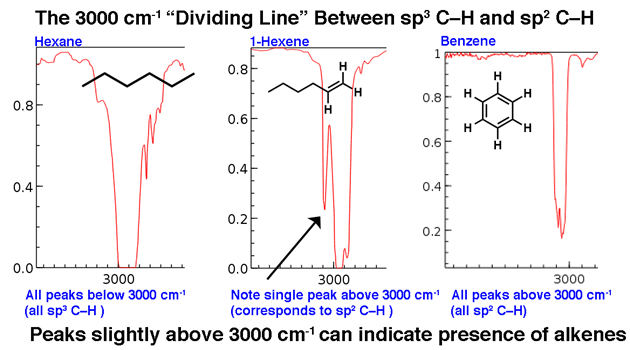
- Die CH-Dehnungsgrenze bei 3000 cm-1
3000 cm & supmin; ¹ dient als nützliche Trennlinie. Oberhalb dieser Linie werden höherfrequente C-H-Strecken beobachtet, die wir sp2-hybridisierten C-H-Bindungen zuschreiben. Zwei Beispiele unten: 1-Hexen (beachten Sie den etwas höheren Peak) und Benzol.
Bei einem Molekül mit nur sp3-hybrisierten CH-Bindungen erscheinen die Linien wie bei Hexan unter 3000 cm-1 , unten.
2. Die markante Dreifachbindungsregion um 2200 cm & supmin; ¹ Moleküle mit Dreifachbindungen erscheinen im großen Schema der Dinge relativ selten, aber wenn sie dies tun, haben sie im IR eine charakteristische Spur.
Die Region zwischen 2000 cm-1 und 2400 cm-1 ist in IR-Spektren eine Art „Geisterstadt“. In dieser Region erscheint nur sehr wenig. Wenn Sie in dieser Region Spitzen sehen, ist dies wahrscheinlich ein dreifach gebundener Kohlenstoff wie ein Alkin oder Nitril.
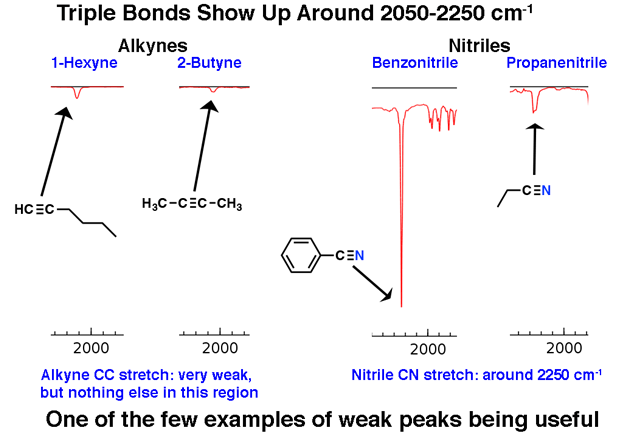
Beachten Sie, wie schwach die Alkinpeaks sind. Dies ist eine Ausnahme von Die Regel, dass man schwache Peaks ignorieren sollte. Dennoch ist Vorsicht geboten: Wenn Sie die Summenformel erhalten, bestätigen Sie, dass ein Alkin möglich ist, indem Sie den Grad der Ungesättigtheit berechnen und sicherstellen, dass er mindestens 2 oder mehr beträgt.
terminale Alkine (wie 1-Hexin) haben ebenfalls eine starke CH-Dehnung um 3400 cm-1, die stärker diagnostisch ist.
Glucose, überarbeitet: Die 1-Minuten-Analyse
OK. Wir sind gegangen r 4 Regionen, die für eine schnelle Analyse eines IR-Spektrums nützlich sind.
Gehen wir nun zurück und betrachten die IR von Glukose. Was sehen wir?
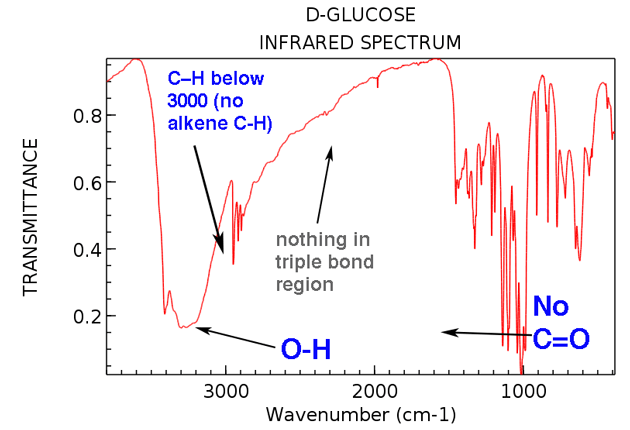
Hier sind die beiden großen Dinge, die zu beachten sind:
Auch wenn Wir nehmen uns etwas mehr Zeit, um zu sehen:
- Kein Alken-CH (keine Peaks über 3000 cm & supmin; ¹)
- Nichts im dreifach gebundenen Bereich (selten, aber immer noch) eine einfache Sache zu lernen zu überprüfen)
Nun: Wenn Sie dieses Spektrum zusammen mit seiner Molekülformel C6H12O6 als „unbekannt“ erhalten würden, welche Schlussfolgerungen könnten Sie über seine Struktur ziehen?
- Das Molekül hat mindestens eine OH-Gruppe (und möglicherweise mehr)
- Das Molekül hat keine C = O-Gruppen
- Das Molekül * wahrscheinlich * hat keine Alkene. Wenn Alkene vorhanden sind, tragen sie keine CH-Bindungen, da sich ihr CH über 3000 cm & supmin; ¹ erstreckt.
A. Molekül mit einem Grad an Wasserstoffmangel (C6H12O6), aber ohne C = O und wahrscheinlich ohne C = C?
Eine gute Vermutung wäre, dass das Molekül einen Ring enthält. (Wir wissen, dass dies der Fall ist, Natürlich, aber es ist schön zu sehen, dass die IR bestätigt, was t wissen wir bereits).
Dies kann uns eine 1-minütige Analyse des IR von Glukose sagen. Wohlgemerkt nicht die gesamte Struktur, aber sicherlich einige wichtige Dinge.
Das reicht für heute. Im nächsten Beitrag werden wir einige weitere 1-minütige Analysen durchführen und konkretere Beispiele dafür geben, wie die Informationen in einem IR-Spektrum verwendet werden können, um Schlussfolgerungen über die Molekülstruktur zu ziehen.
Mehr zur 3200-Region: Amine , Amide und Terminal Alkyne CH
Während wir uns in der Region 3200 befinden…. Amine und Amide
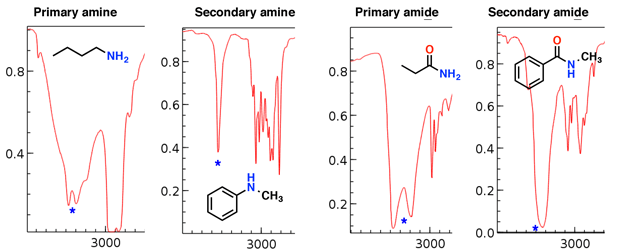
Amine und Amide weisen ebenfalls N-H-Abschnitte auf, die in dieser Region auftreten.
Beachten Sie, dass das primäre Amin und das primäre Amid zwei „Reißzähne“ aufweisen, während das sekundäre Amin und das sekundäre Amid einen einzelnen Peak aufweisen.
Die Aminstrecken sind tendenziell schärfer als das Amid Dehnungen, auch die Amide können durch eine starke C = O-Dehnung unterschieden werden (siehe unten).
Primäre Amine (Klicken für Spektren)
- Anilin
- Benzylamin
- Cyclohexylamin
Sekundäre Amine:
- N-Methylbenzylamin
- N, N-Dibenzylamin
- N-Methylanilin
Primäre Amide
- Propionamid
- Benzamid
- Butanamid
Sekundäre Amide
- N-Methylbenzamid
terminales Alkin CH
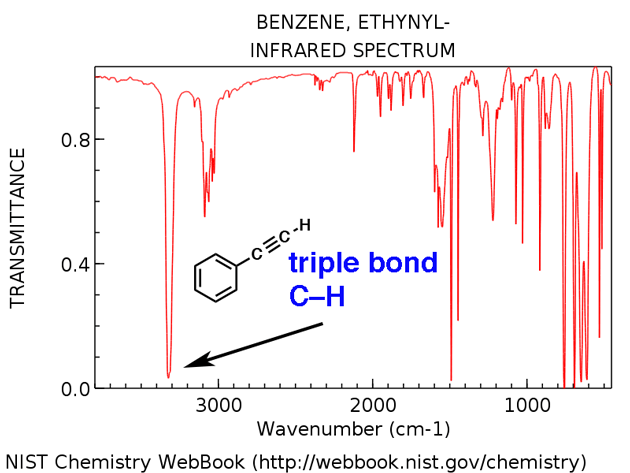
terminale Alkine haben eine charakteristische CH-Dehnung um 3300 cm & supmin; ¹. Hier ist es unten für Ethinylbenzol.
- Ethinylbenzol