Hur man tolkar IR Spectra på 1 minut eller mindre: De 2 viktigaste sakerna att leta efter
Senaste inlägget introducerade vi kort begreppet bindningsvibrationer och vi såg att vi kan tänka på kovalenta bindningar som lite som kulor och fjädrar: fjädrarna vibrerar och var och en ”sjunger” med en karakteristisk frekvens , som beror på bindningens styrka och på atommassorna. Dessa vibrationer har frekvenser som ligger i det infraröda området (IR) i det elektromagnetiska spektrumet.
Vi kan observera och mäta detta ”sjunga” bindningar genom att applicera IR-strålning på ett prov och mäta de frekvenser vid vilka strålningen absorberas. Resultatet är en teknik som kallas infraröd spektroskopi, vilket är ett användbart och snabbt verktyg för att identifiera bindningarna i en given molekyl.
Vi såg att IR-spektrumet för vatten var ganska enkelt – men att gå vidare till en relativt komplex molekyl som glukos (nedan) konfronterades vi plötsligt med en skog av toppar!
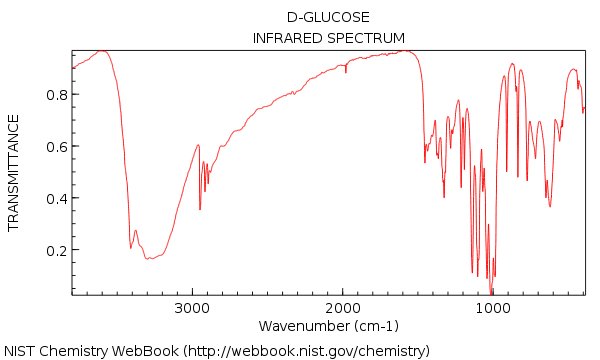
Ditt första intryck av att se vid den IR kan vara: agh! hur ska jag förstå det ??
Till vilket jag vill säga: kom inte i panik!
Innehållsförteckning
- Låt oss korrigera några vanliga missuppfattningar om IR
- Att börja med ”Hunt And Peck” är inte vägen att gå
- IR-spektroskopi: den stora bilden
- De två viktigaste sakerna att leta efter i ett IR-spektrum: ”tungor” och ”svärd”.
- Alkoholer och karboxylsyror: Mer Detalj
- Specifika exempel på IR-spektra för karbonylfunktionella grupper
- Mindre avgörande, men ändå användbar: Ytterligare två mycket diagnostiska områden.
- Glukos, Revisited: The 1 Minutanalys
Låt oss korrigera några vanliga missuppfattningar om IR
I det här inlägget vill jag visa att en typisk analys av ett IR-spektrum är mycket enklare än du kanske tror Faktum är att när du väl har lärt dig vad du ska leta efter kan det ofta göras på en minut eller mindre. Varför?
- IR används vanligtvis inte för att bestämma hela strukturen i en okänd molekyl. Det finns till exempel inte en person vid liv som kan titta på IR-spektrumet ovan och härleda strukturen för lim kose från det. IR är ett verktyg med en mycket specifik användning.
- Vi behöver inte analysera varje topp! (som vi kommer se senare är det vad NMR är för: -)). Istället är IR bra för att identifiera vissa specifika funktionella grupper, som alkoholer och karbonyler. På det här sättet är det gratis för andra tekniker (som NMR) som inte ger denna information lika snabbt.
Med detta i åtanke kan vi förenkla analysen av ett IR-spektrum genom att klippa ut allt utom den lägst liggande frukten.
Ser du den skog av toppar från 500-1400 cm-1? Vi kommer i princip att ignorera dem alla!
80% av den mest användbara informationen för våra ändamål kan erhållas genom att titta på två specifika områden i spektrumet: 3200-3400 cm-1 och 1650-1800 cm-1. Vi ser också att det finns åtminstone ytterligare två regioner av ett IR-spektrum som är värt att titta på och därmed avslutar en ”första ordning” -analys av ett okänt IR-spektrum.
Slutsats: syftet med det här inlägget är att visa dig hur du prioriterar din tid i en analys av ett IR-spektrum.
2. Att börja med ”Hunt And Peck” är inte vägen att gå
Konfronterat med ett IR-spektrum av okänd (och en känsla av stigande panik), vad gör en typisk ny student?
De når ofta det första verktyget de får, vilket är en tabell över vanliga intervall för IR-toppar som deras instruktör gav dem.
Nästa steg i deras analys är att gå igenom spektrumet från en sida till nästa och försöka matcha varje topp till ett av siffrorna i tabellen . Jag vet detta eftersom det var precis vad jag gjorde när jag först lärde mig IR. Jag kallar det ”jakt och plockning”.
De enda människorna som ”jagar och plockar” som sitt första steg är människor som inte har någon plan (dvs ”nybörjare”).
Så genom att läsa de närmaste styckena kan du spara mycket tid och förvirring.
Den stora bilden
I IR-spektroskopi mäter vi var molekyler absorberar fotoner av IR-strålning. Topparna representerar områden i spektrumet där specifika bindningsvibrationer förekommer. Precis som fjädrar med olika vikter vibrerar vid karakteristiska frekvenser beroende på massa och spänning, så gör bindningar också.
Här är en översikt över IR-fönstret från 4000 cm -1 till 500 cm -1 med olika intressanta regioner markerade.
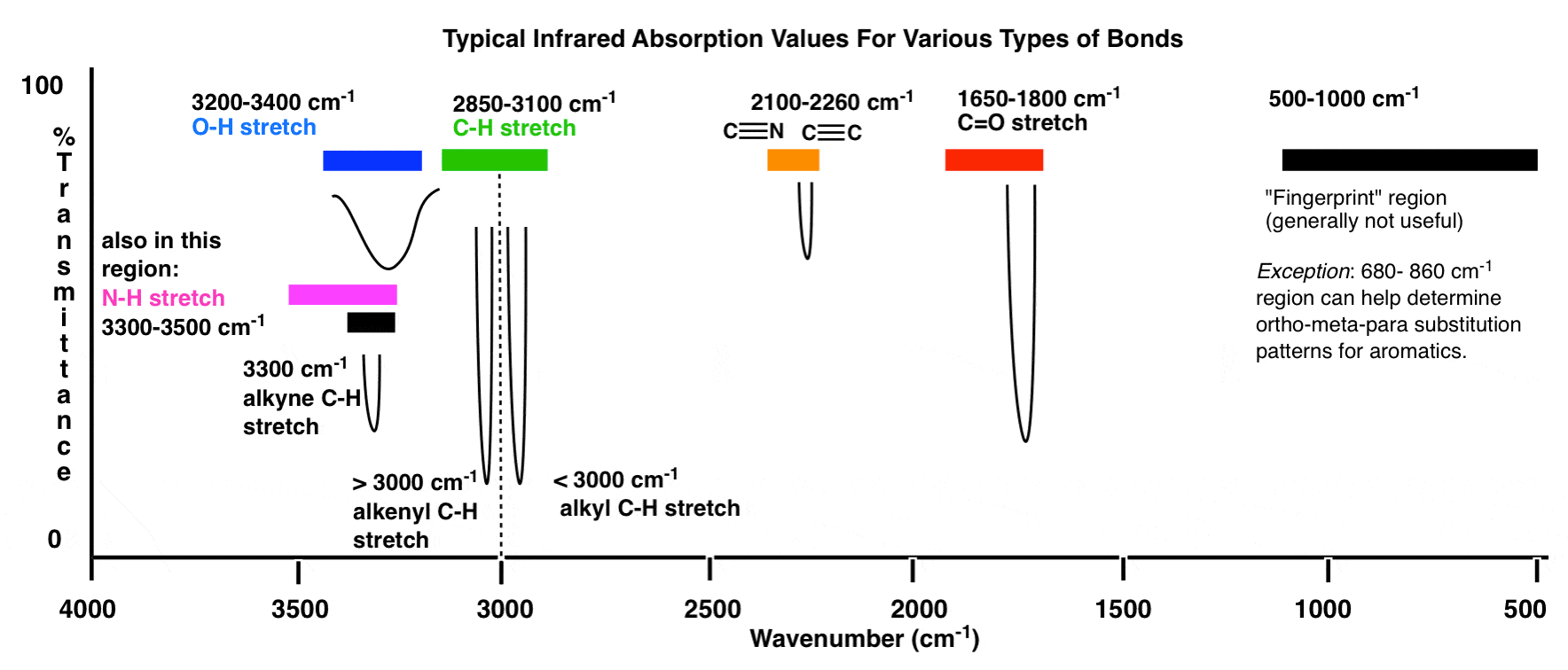
En ännu mer komprimerad översikt ser ut så här: (källa)
Inom dessa intervall finns det två högprioriterade områden att fokusera på och två mindre prioriterade områden som vi diskutera vidare nedan.
4. De två viktigaste sakerna att leta efter i ett IR-spektrum: ”Tungor” och ”Svärd”.
När du står inför ett nytt IR-spektrum, prioritera din tid genom att ställa två viktiga frågor:
- Finns det en bred, rundad topp i regionen runt 3400-3200 cm- 1? Det är där hydroxylgrupper (OH) dyker upp.
- Finns det en skarp, stark topp i regionen omkring 1850-1630 cm-1? Det är där karbonylgrupper (C = O) dyker upp.
Låt oss först titta på några exempel på hydroxylgruppstoppar i regionen 3400 cm-1 till 3200 cm-1, som Jon beskriver levande som ”tungor”. Topparna nedan hör till alkoholer. Vätebindning mellan hydroxylgrupper leder till vissa variationer i OH-bindningsstyrka, vilket resulterar i en rad vibrationsenergier. Variationen resulterar i de observerade breda topparna.
Hydroxylgrupper som ingår i karboxylsyror har ett ännu bredare utseende som vi kommer att beskriva lite.
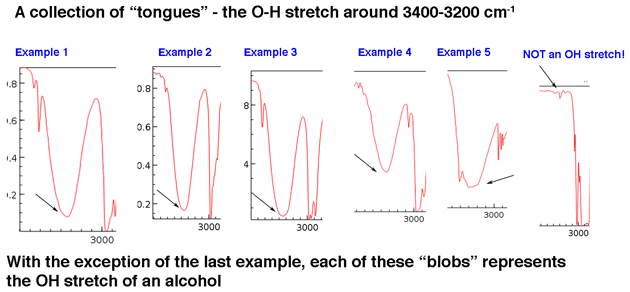
Huvudpunkten är att en hydroxylgrupp i allmänhet inte är något du behöver leta efter i basljudet.
Även om hydroxylgrupper är den vanligaste typen av bred topp i denna region, toppar NH kan också visas i detta område (mer om dem i fotnot nedan). De tenderar att ha ett skarpare utseende och kan se ut som en eller två toppar beroende på antalet NH-bindningar.
Nästa, le t tittar på några exempel på C = O-toppar, i regionen omkring 1630-1800 cm-1 .. Dessa toppar är nästan alltid de starkaste topparna i hela spektrumet och är relativt smala, vilket ger dem ett något ”svärdliknande” utseende .
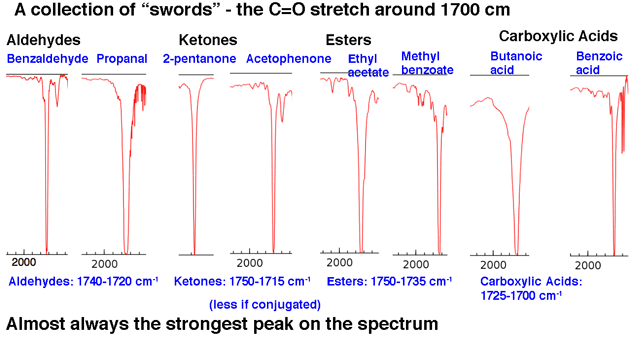
Det sammanfattar vår 80/20 analys: leta efter tungor och svärd.
Om du lär dig ingenting annat från det här inlägget, lär dig att känna igen dessa två typer av toppar!
Två andra regioner i IR-spektrumet kan snabbt ge användbar information om du tränar dig själv att leta efter dem.
3. Linjen vid 3000 cm-1 är en användbar ”gräns” mellan alken C – H (över 3000 cm-1) och alkan C – H (under 3000 cm-1) Detta kan snabbt hjälpa dig att avgöra om dubbelbindningar finns.
4. En topp i regionen runt 2200 cm-1 – 2050 cm-1 är en subtil indikator på närvaron av en trippelbindning. Inget annat dyker upp i denna region.
En påminnelse om sunt förnuft
Först några uppenbara råd:
- om du får molekylformeln, kommer det att avgöra vilka funktionella grupper du ska leta efter. Det gör ingen meningsfullt att leta efter OH-grupper om du inte har några oxygener i din molekylformel, eller på samma sätt närvaron av en amin om formeln saknar kväve.
- Mindre uppenbart beräknar du omättnadsgraden om du får molekylen formel, eftersom det kommer att ge viktiga ledtrådar. Leta inte efter C = O i en struktur som C4H10O som inte har några grader av omättnad.
5. Alkoholer och karboxylsyror: Mer detaljer
Alkoholer
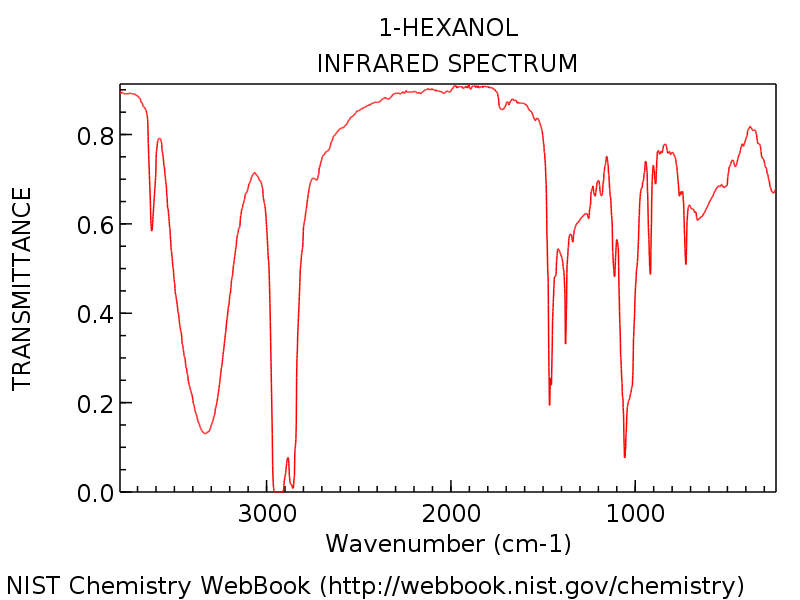
Le Vi tittar på ett specifikt exempel så att vi kan se allt i perspektiv. Spektrumet nedan består av 1-hexanol.
Observera hydroxylgruppstoppen runt 3300 cm-1, typisk för en alkohol (den skarpa toppen runt 3600 cm-1 är en vanlig följeslagare till hydroxyltoppar: den representerar icke vätebunden OH).
Som du kan förvänta dig för 1-hexanol finns det ingen uppenbar karbonyltopp runt 1700 cm-1. Nybörjare kan vara frestade att märka den dolkliknande starka toppen vid cirka 1450 cm-1 som en möjlig C = O-sträcka. Det är det inte. (det är förmodligen en C-H-böjning). Variationer förekommer bara inom ett mycket smalt intervall, och det är mycket osannolikt att du ser en C = O-sträcka mycket under 1650 cm-1. Ju fler spektra du ser, desto bättre blir du att göra dessa bedömningar.
För att få lite förtrogenhet med variationen, här är några fler exempel av hela IR-spektra av olika alkoholer.
- Fenol
- Cyklohexanol
- 1-butanol
Karboxylsyror
Hydroxylgrupper i karboxylsyror är betydligt bredare än i alkoholer. Jon kallar det ett ”hårigt skägg”, vilket är en perfekt beskrivning. Deras utseende är också mycket varierande. OH-absorptionen i karboxylsyror kan vara så bred att den sträcker sig under 3000 cm-1, ganska mycket ”tar över” vänster hand del av spektrumet.
Här är ett exempel: butansyra.
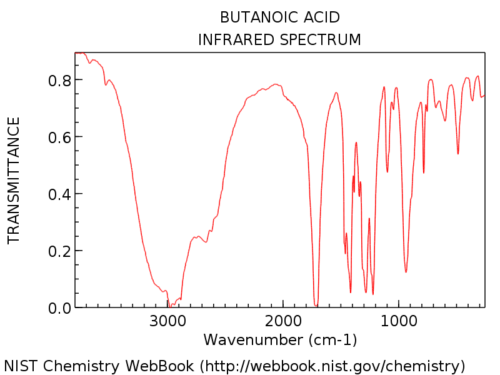
Här är några fler exempel på fulla spektra så att du kan se variationen.
- bensoesyra,
- pentansyra,
- ättiksyra
Skillnaden i utseende mellan OH för en alkohol och en karboxylsyra är vanligtvis diagnostisk. I det sällsynta fall där du inte är säker på om den breda toppen beror på OH av en alkohol eller en karboxylsyra, är ett förslag att kontrollera området runt 1700 cm för C = O-sträckan. Om den saknas tittar du sannolikt på en alkohol.
Specifika exempel på IR-spektra av karbonylfunktionella grupper
Den andra viktiga toppregionen är karbonyl C = O-sträckningsområdet ungefär 1630-1830 cm. Karbonylsträckor är skarpa och starka.
När du ser några av dem är de omöjliga att missa. Inget annat dyker upp i denna region.
För att sätta det i perspektiv, här är IR-spektrumet av hexanal. Den toppen strax efter 1700 cm-1 är C = O-sträckan. När den är närvarande är C = O-sträckan nästan alltid den starkaste toppen i IR-spektrumet och omöjlig att missa.
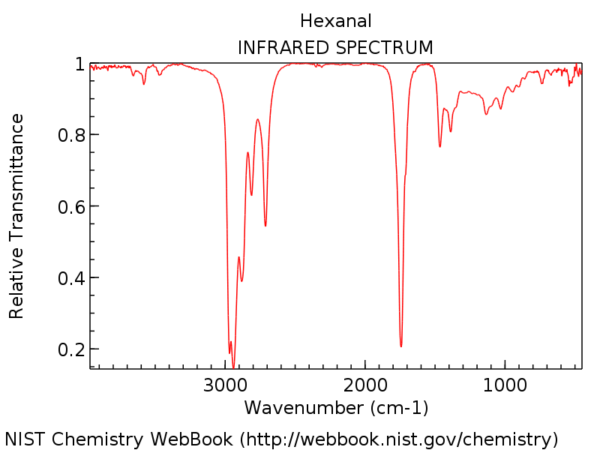
Positionen för C = O-sträckningen varierar något efter karbonylfunktionell grupp. Vissa intervall (i cm-1) visas nedan:
Böjning påverkar positionen för C = O-sträckningen något och flyttar den till lägre vågnummer.
En anständig tumregel är att du aldrig någonsin kommer att se en C = O-sträcka under 1630. Om du till exempel ser en stark topp vid 1500 är det inte C = O. Det är något annat.
Mindre avgörande, men ändå användbart: Två mer mycket diagnostiska områden.
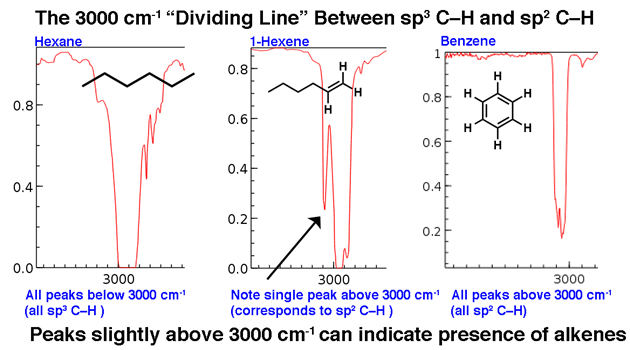
- CH-sträckgränsen vid 3000 cm-1
3000 cm-1 fungerar som en användbar skiljelinje. Över denna linje observeras C-H-sträckningar med högre frekvens som vi tillskriver SP2-hybridiserade CH-bindningar. Två exempel nedan: 1-hexen (notera toppen som står lite högre) och bensen.
För en molekyl med endast sp3-hybriiserade CH-bindningar visas linjerna under 3000 cm-1 som i hexan , nedanför.
2. Den distinkta trippelbindningsregionen runt 2200 cm-1
Molekyler med trippelbindningar förekommer relativt sällan i det stora schemat av saker, men när de gör det har de ett distinkt spår i IR.
Regionen mellan 2000 cm-1 och 2400 cm-1 är lite av en ”spökstad” i IR-spektra. Det finns väldigt lite som visas i denna region. Om du ser toppar i denna region är en trolig kandidat ett trippelbundet kol, såsom en alkyn eller nitril.
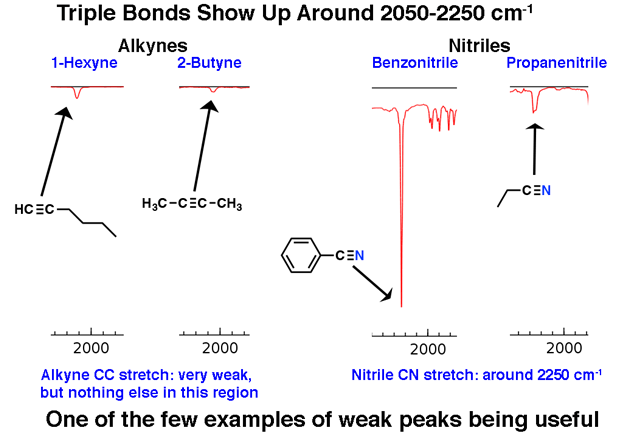
Observera hur svaga alkyntopparna är. Detta är ett undantag från regeln att man ska ignorera svaga toppar. Ändå krävs försiktighet: om du får molekylformeln, bekräfta att en alkyn är möjlig genom att beräkna graderna av omättnad och se till att den är minst 2 eller mer.
Terminalalkyner (som 1-hexyn) har också en stark CH-sträcka runt 3400 cm-1 som är starkare diagnostisk.
Glukos, återbesökt: 1-minutsanalysen
OK. Vi har gått r 4 regioner som är användbara för en snabb analys av ett IR-spektrum.
Låt oss gå tillbaka och titta på IR för glukos. Vad ser vi?
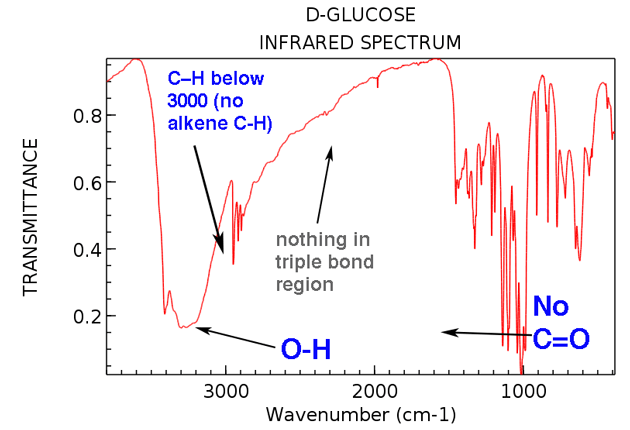
Här är de två stora sakerna att notera:
Även om vi tar lite extra tid vi kan se:
- Ingen alken CH (inga toppar över 3000 cm-1)
- Ingenting i trippelbunden region (sällsynt, men ändå en lätt sak att lära sig att kontrollera)
Nu: Om du fick detta spektrum som ett ”okänt” tillsammans med dess molekylformel, C6H12O6, vilka slutsatser kan du dra om dess struktur? / p>
- Molekylen har minst en OH-grupp (och möjligen fler)
- Molekylen har inga C = O-grupper
- Molekylen * troligtvis * har inga alkener. Om det finns några alkener har de inga CH-bindningar, för vi skulle se deras CH sträcka sig över 3000 cm-1.
A molekyl med en grad av vätebrist (C6H12O6) men ingen C = O och troligen ingen C = C?
En bra gissning skulle vara att molekylen innehåller en ring. (Vi vet att detta är fallet, naturligtvis, men det är trevligt att se IR bekräfta vem Vi vet det redan).
Det här är vad en 1-minuts analys av glukos IR kan berätta för oss. Inte hela strukturen, tänk på det, men verkligen några viktiga bitar.
Det räcker för idag. I nästa inlägg gör vi några fler analyser på 1 minut och ger mer konkreta exempel på hur man använder informationen i ett IR-spektrum för att dra slutsatser om molekylär struktur.
Mer om 3200-regionen: Aminer , Amides och Terminal Alkyne CH
Medan vi är i 3200-regionen …. Aminer och amider
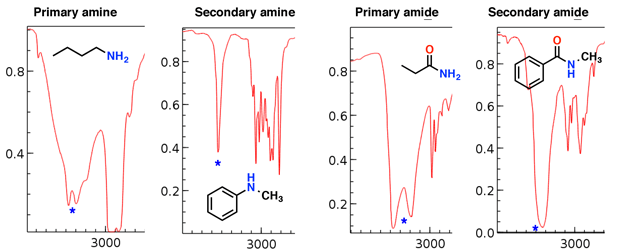
Aminer och amider har också N-H-sträckor som dyker upp i denna region.
Lägg märke till hur den primära aminen och den primära amiden har två ”huggtänder”, medan den sekundära aminen och den sekundära amiden har en enda topp.
Aminsträckorna tenderar att vara skarpare än amiden sträckor; även amiderna kan särskiljas med en stark C = O-sträcka (se nedan).
Primära aminer (klicka för spektra)
- Aniline
- Bensylamin
- Cyklohexylamin
Sekundära aminer:
- N-metylbensylamin
- N, N-dibensylamin
- N-metylanilin
Primära amider
- Propionamid
- Bensamid
- Butanamid
Sekundära amider
- N-metylbensamid
Terminalalkyn CH
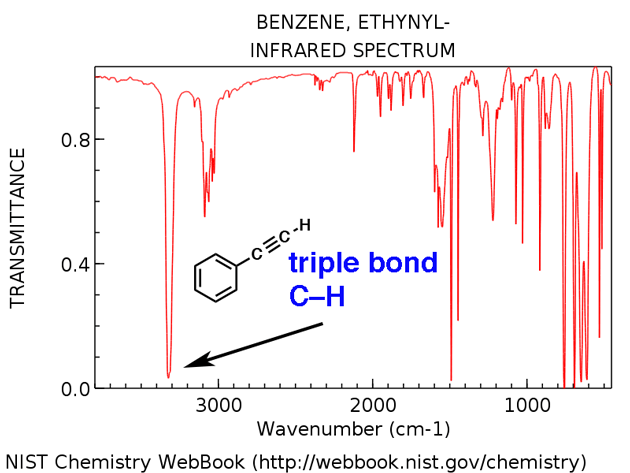
Terminalalkyner har en karakteristisk CH-sträckning runt 3300 cm-1. Här är det för etynylbensen, nedan.
- Etynylbensen